Case Report
Urethral Duplications in Children – A Case Series
Vuille-dit-Bille RN1,2*#, Leu S3#, R. Delcont M4, Ulf Kessler5, Zeino M5 and Klimek P1
1Department of Pediatric Surgery, Cantonal Hospital of Aarau, Switzerland
2Department of Pediatric Surgery, Children’s Hospital Colorado, USA
3Department of Pediatric Surgery, University of Zurich, Switzerland
4Department of Pediatric Surgery, University of Colorado School of Medicine, USA
5Department of Pediatric Surgery, Bern University Hospital, Switzerland
#Contribute equally for the manuscript
*Corresponding author: Raphael N Vuille-dit-Bille, Department of Pediatric Surgery, Children’s Hospital Colorado, 13123 E 16th Ave, Aurora, CO 80045, USA
Published: 14 Sep, 2018
Cite this article as: Vuille-dit-Bille RN, Leu S, R. Delcont M,
Ulf Kessler, Zeino M, Klimek P. Urethral
Duplications in Children – A Case
Series. Clin Surg. 2018; 3: 2099.
Abstract
Background: Urethral duplications are rare congenital anomalies with multiple anatomical variants.
They mostly occur in the sagittal plane and can be associated with other congenital urogenital
malformations. According to Effmann urethral duplications are classified into incomplete (type I),
complete (type II), and complete associated with caudal duplication (type III).
Methods: We hereby describe three cases of urethral duplications and their respective treatment.
Results: Two cases were type I duplications. One patient was treated by complete resection of the
blind ending secondary urethra, whereas no treatment was performed in the second patient. The
third case was a IIA-2 Y-subtype duplication that needed complex reconstruction.
Conclusions: There is no need for surgical management in asymptomatic patients with type I
urethral duplications. Type IIA-2 Y-subtype duplications are extremely rare, and their treatment
must be individualized according to anatomical and physiological features.
Keywords: Urethral duplication; Y-duplication; Urethral anomaly
Introduction
Urethral duplications are seldomly seen urinary tract variants consisting of aberrant partial or
complete development of an additional urethra [1]. They may be associated with different urogenital
abnormalities, including bladder duplication and bladder exstrophy [2]. The etiology of this disease
is not well understood. In the majority of cases, urethral duplications occur in the sagittal plane with
the accessory urethra located dorsally to the orthotopic urethra [3]. The most commonly applied
classification according to Effmann et al. divides urethral duplications into incomplete duplications
(type I), complete duplications (type II), and duplications as a component of partial or complete
caudal duplication (type III) [4]. Types I and II are further subdivided (Table 2) [5]. Whereas Type
I duplications are typically clinically silent without urinary discharge or infections of the accessory
urethra, complete duplications may be the cause for urinary incontinence and repeat urinary tract
infections.
The aim of this case series is to describe the clinical presentation and respective treatment of this
rare disease in three male children suffering from different types of urethral duplication.
Materials and Methods
We hereby present three male patients born between September 2008 and November 2013 with urethral duplications (Table 1). Thereof, two children had type IA duplications according to Effmann et al [4]. In one child, the ectopic urethra was successfully excised, whereas there was no treatment in the other child who also had Down syndrome. The third child presented with IIA- 2 Y-subtype urethral duplication which consisted of a dystopic urethral orifice into the anus and a dysplastic dorsal urethra. A three-staged procedure was performed to restore the anatomy. All children were treated at two pediatric surgery centers in Switzerland (the University Hospital of Bern and the Cantonal Hospital of Aarau).
Figure 1
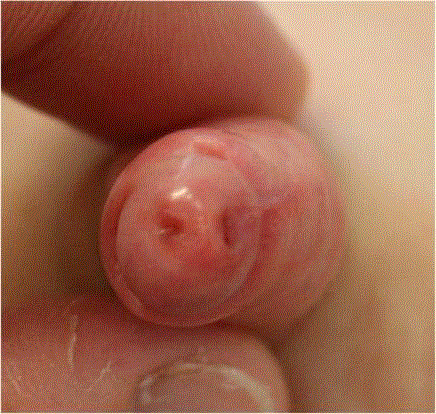
Figure 1
Type 1A urethral duplication. Unrepaired blind ending secondary
orifice in the axial plane lateral to the orthotopic urethra.
Figure 2
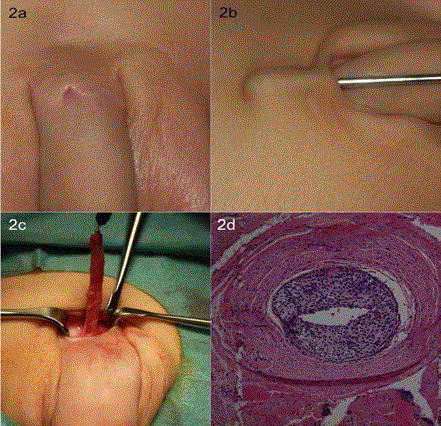
Figure 2
Type 1A urethral duplication. (A) Preoperative image of accessory
pore originating 2 cm proximal to the orthotopic urethra. (B) Sounding of
pore and marking of secondary urethra with betadine injection. (C) Complete
accessory urethral resection (D) Histology showing fistula tract with
urothelium and low-grade chronic inflammation.
Results
Case 1
Case 1 describes a boy with Down syndrome who suffered from
spontaneously resolving Myelodysplastic Syndrome (MDS) in the
neonatal period. A secondary urethral orifice was found incidentally
by the child’s paediatric oncologist at 5 years old (Figure 1). No
urinary tract or local infections were noted. Ultrasound showed an
unaffected bladder without wall thickening, as well as proportionately
sized and normally structured kidneys and ureters. Micturating
cystourethrogram demonstrated an anatomically positioned penile
urethra arising from the common bladder neck. No vesicoureteral
reflux was noted, and the bladder could be fully emptied. The
secondary orifice was blind ending without connecting to the primary
urethra or the bladder, corresponding to Type IA (Table 2). Because
it was a short, incomplete and hence asymptomatic duplication,
surgical excision was not performed in accordance with the child’s
mother.
Case 2
Case 2 describes a boy with a blind-ending accessory urethral
orifice incidentally discovered at an inguinal hernia repair follow-up
when he was 10 months old (Figure 2A). The boy was asymptomatic,
without discharge of urine from the accessory orifice, and otherwise
healthy. Ultrasound revealed a blind ending secondary urethra 2 cm
proximal to the orifice, along with unaffected intra and retroperitoneal
organs. Eleven months after first recognition, complete circumcision
due to symptomatic phimosis in combination with excision of the
secondary urethra was performed. The pore was sounded (Figure 2B),
and betadine solution was injected to mark the course of the secondary
urethra. The pore was closely cut down to the orthotopic urethra and
resected completely (Figure 2C). The operation was performed in an
outpatient setting. Histologic examination confirmed a fistula tract
with urothelium and low-grade chronic inflammation (Figure 2D).
Postoperative follow-up 1.5 months after excision showed complete
wound healing without relapse.
Case 3
Case 3 presented as a newborn boy with severe pyelonephritis.
The patient passed urine rectally with only a few drops from the dorsal
glandular meatus (Figure 3A), concomitant with IIA-2 Y-subtype
urethral duplication. Further diagnostic work-up (renal ultrasound,
micturating cystourethrogram, Tc-99m dimercaptosuccinic acid
renal scintigraphy, and magnetic resonance urography) showed
right-sided grade 4 vesicoureteral reflux with 10% function of the
right kidney. Unilateral right-sided loop cutaneous ureterostomy
[6] was performed without complication at 3 months old allowing
retrograde urination from the bladder, and the child was discharged
4 days later after an uneventful post-operative course.
Separation and mobilization of the ventral urethra was
performed at 14 months old using an anterior sagittal trans-anorectal
approach. The hypoplastic distal urethra was resected, and urethral
bed substitution using a pedicle preputial flap (first stage of the twostage
repair according to Bracka [7]) was performed (Figure 3B).
Surgery was uneventful. At 1.5 years old, the second stage of urethral
reconstruction, consisting of tubularization of the graft, was done
together with right-sided nephroureterectomy due to worsening of
right-sided kidney function to less than 10% (Figure 3C and 3D).
The scrotum bifidum was also corrected with scrotal transposition,
a urethral stent was inserted, and a suprapubic catheter was placed.
The patient was anuric during the immediate postoperative
period, so emergent abdominal ultrasound was performed. This
showed an intraperitoneal urinoma together with pyelectasis of the
remaining left kidney. Emergent reoperation revealed a left-sided
ureteral injury by virtue of crossed dystopia of the kidney (left to
right) that was not diagnosed before this operation. The ureter was
mobilized to the left side, and ureteroneocystostomy using the psoas
hitch ureteral reimplantation technique [8] was undertaken. Voiding
cystourethrogram performed 18 days after the second stage of
urethral reconstruction showed a small leakage at the middle section
of the penile urethra. The suprapubic catheter was hence left in
place. Repeat voiding cystourethrogram one week thereafter showed
no fistula and a normal sized urethra, so the suprapubic catheter
was removed. The subsequent five-month follow-up showed good
cosmetic and functional results without any complications.
Figure 3
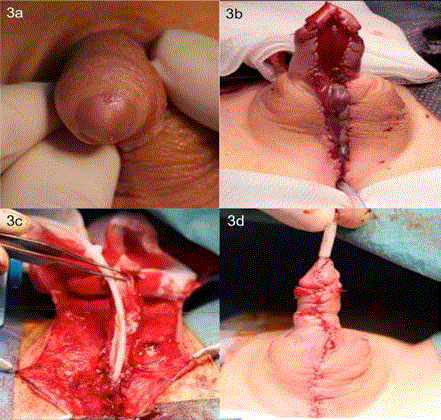
Figure 3
Urethral duplication IIA-2 Y-subtype repair. (A) Preoperative
picture of hypoplastic, nonfunctional dorsal glandular meatus. (B) Resection
of hypoplastic distal urethra and urethral bed substitution using a pedicle
preputial flap, according to Bracka et al [7]. (C) Tubularization of graft,
insertion of urethral stent, and correction of scrotum bifidum with scrotal
transposition (D) completed urethroplasty.
Discussion
Urethral duplication reflects a very rare urogenital malformation
that occurs more frequently in males than females. Only a few more
than 325 cases have been reported in the literature to date [9]. They
typically occur in the sagittal plane [10] and consist of squamous
epithelia surrounded by smooth muscle identical to the orthotopic
urethra. The aetiology of urethral duplication is still not clear
and is attributed to altered embryogenesis. The most commonly
quoted hypothesis is the one of Patten and Barry, which purports
a misalignment of the lateral folds of the genital tubercle and the
ventral end of the cloacal membrane. Other authors describe urethral
duplications as due to Mullerian duct termination and growth arrest of
the urogenital sinus [10]. However, no single theory is able to explain
all different subtypes of urethral duplication. Different anatomical
variants of urethral duplication led to several different classification
systems [11], with that proposed by Effmann and colleagues being
the most used [3]. Effmann divides urethral duplications into three
different types (Table 2). In a series of pediatric urology case reports,
the relative frequency of type I, type II, and type III duplications was
25%, 63%, and 12%, respectively [9]. In the present case series, two
type IA cases with blind ending accessory urethras and one IIA-2
Y-subtype are shown.
Most blind-ending accessory urethras are clinically asymptomatic
and rarely associated with other anomalies. If symptoms exist, they
may include sinus tract infection and mucus discharge. One child
with type IA duplication in the present study was otherwise healthy,
while the other child had Down syndrome. Both duplications
were incidentally noted by health professionals during physical
examination. There’s no need for therapeutic management in
asymptomatic patients apart from cosmetic issues. Accordingly, the
child with type IA duplication and symptomatic phimosis was treated
by complete excision of the blind ending urethra, whereas the other
child with Down syndrome was not treated.
In contrast to type I duplications, type II and type III duplications
are associated with clinical symptoms including a doubled urinary
stream, urinary incontinence, recurrent urinary tract infections,
bladder outflow tract obstruction, and passing urine per anus with
voiding [3,9,12-14]. Type II and III duplications occur along with
more severe congenital anomalies. A variant of type IIA-2 duplication,
referred to as the ‘IIA-2 Y-subtype’, describes a secondary urethra
coursing into a second meatus that opens posteriorly into the rectum/
perineum [1]. This type is extremely rare with less than 20 cases being
reported until the year 2000 and is associated with hypospadias,
epispadias, cleft palate, congenital heart disease, tracheoesophageal
fistula, and imperforate anus [9,10,14]. Type III, which describes
complete or partial caudal urethral duplication, occurs most often
with bladder and/or penis duplication, as well as the afore-mentioned
abnormalities [3].
These duplications (type II and III) are hence typically diagnosed
directly at birth [15]. Correspondingly, the child presenting with the
IIA-2 Y-subtype duplication in this study showed repeated urinary
tract infections, as well as higher grade vesicoureteral reflux with
reduced kidney function. Surgical treatment was necessary to relieve
the kidney and to restore anatomy. A three-step procedure was thereby
chosen consisting of (i) unilateral loop cutaneous ureterostomy at 3
months old, (ii) separation and mobilization of the ventral urethra
at 14 months old, and (iii) urethral reconstruction at 18 months old.
Conclusion
In general, there is no need for therapeutic management in asymptomatic patients with type I urethral duplications apart from cosmetic issues. Type IIA-2 Y-subtype duplications are extremely rare, and their treatment must be individualized according to anatomical and physiological features. Vesicoureteral reflux and repeated urinary tract infections are common co-findings of this subtype of urethral duplication.
Funding
This work was supported by the Research Council Cantonal Hospital Aarau [Grant 1410.000.076 to P.K., R.N.V., and M.Z].
References
- Alanee S, Gupta P, Gleich P, Shukla AR. Complete urethral duplication: Description of surgical approach mimicking distal epispadias repair. J Pediatr Urol. 2012;8(4):343-7.
- Baid M, Dutta A. Urethral duplication in a 15-year-old: case report with review of the literature. Rev Urol. 2014;16(3):149-51.
- Bracka A. A versatile two-stage hypospadias repair. Br J Plast Surg. 1995;48(6):345-52.
- Coleman RA, Winkle DC, Borzi PA. Urethral duplication: Cases of ventral and dorsal complete duplication and review of the literature. J Pediatr Urol. 2010;6(2):188-91.
- Gupta NP, Ansari MS, Aron M, Mandal S. Y duplication of urethra with complete atresia of the orthotopic channel: 1-stage reconstruction. J Urol. 2000;163(3):949-50.
- Haleblian G, Kraklau D, Wilcox D, Duffy P, Ransley P, Mushtaq I. Y-type urethral duplication in the male. BJU Int. 2006;97(3):597-602.
- Levin TL, Han B, Little BP. Congenital anomalies of the male urethra. Pediatr Radiol. 2007;37(9):851-62.
- Manassero F, Mogorovich A, Fiorini G, Di Paola G, De Maria M, Selli C. Ureteral reimplantation with psoas bladder hitch in adults: A contemporary series with long-term followup. Scientific World J. 2012;379316.
- Mane SB, Obaidah A, Dhende NP, Arlikar J, Acharya H, Thakur A, et al. Urethral duplication in children: our experience of eight cases. J Pediatr Urol. 2009;5(5):363-7.
- Mori K, Shin T, Tobu S, Noguchi M, Sumino Y, Sato F, et al. A case of urethral duplication arising from the posterior urethra to the scrotum with urinary stone in a 6-year-old male. Case Rep Pediatr. 2014;290623.
- Onofre LS. Urethral duplication--a wide spectrum of anomalies. J Pediatr Urol. 2013;9(6, Pt B):1064-71.
- Podesta ML, Medel R, Castera R, Ruarte AC. Urethral duplication in children: surgical treatment and results. J Urol. 1998;160(5):1830-3.
- Singh S, Rawat J. Y-type urethral duplication in children: Management strategy at our center. J Indian Assoc Pediatr Surg. 2013;18(3):100-4.
- Wagner JR, Carr MC, Bauer SB, Colodny AH, Retik AB, Hendren WH. Congenital posterior urethral perineal fistulae: A unique form of urethral duplication. Urology. 1996;48(2):277-80.
- Zilberman DE, Golomb J, Kitrey ND, Inbar Y, Heyman Z, Kleinnbaum Y, et al. Long-term urinary bladder function following unilateral refluxing low loop cutaneous ureterostomy. Korean J Urol. 2012;53(5):355-9.