Case Report
The Evolving Genetic Landscape of Hirschprung Disease: An Update and Review
Amit Kumar Yadav* and Gaurav Chopra
Department of Pathology, Vardhman Mahavir Medical College and Safdarjung Hospital, New Delhi, India
*Corresponding author: Amit Kumar Yadav, Department of Pathology, Vardhman Mahavir Medical College and Safdarjung Hospital, New Delhi, 110029, India
Published: 11 Oct, 2017
Cite this article as: Yadav AK, Chopra G. The Evolving
Genetic Landscape of Hirschprung
Disease: An Update and Review. Clin
Surg. 2017; 2: 1672.
Abstract
Hirschsprung Disease (HD) is a developmental disorder characterized by the complete absence
of ganglion cells in the distal gastrointestinal tract. It is the most common cause of functional
intestinal obstruction in neonates and children. The aganglionosis is believed to be either due to
failure of neural crest cells to migrate, proliferate, differentiate or survive during gut development
in the embryonic stage. The incidence of HD is estimated at 1/5000 live births and shows a male
predominance. It is usually sporadic, although it can be familial and may be inherited as autosomal
dominant or autosomal recessive. In 70% of cases, HD occurs as an isolated trait and in the
other 30% it is associated with other congenital malformation syndromes. HD has a complex
multifactorial etiology, and genetic factors play a key role in its pathogenesis. Several gene loci
appear to be involved. Many of these have been identified by conducting Genome Wide Association
(GWAS) studies and recently by Next Generation Sequencing (NGS). These genes encode for
receptors, ligands (especially those participating in the RET, EDNRB and Semaphorin signaling
pathways), transcriptional factors (PHOX2B & SOX10). These genes are involved in the neural crest
cell development and migration that give rise to ganglion cells. Overall, the RET proto-oncogene is
considered the major disease causing gene in HD. A greater understanding of the genetic landscape
of this disease might pave way for genetic counseling, prenatal and preimplantation diagnosis in the
management of HD.
Keywords: Hirschprung disease; Genetics; RET; Genome wide association studies (GWAS);
Next generation sequencing (NGS); Semaphorins
Introduction
Hirschsprung Disease (HD), or congenital intestinal aganglionosis, is a pediatric disorder which
is characterized by complete absence of ganglion cells from a part of the intestine. Its incidence is
reported to be 1:5000 births [1]. The neuronal ganglion cells are of neural crest origin. Since during
development the migration of these cells is from proximal to distal the aganglionic segment typically
includes the distal rectum and proximal extent is variable. The absence of ganglion cells has been
attributable to either failure of migration of these enteric neural crest cells or due to absence of
survival, proliferation or differentiation of these cells [2].
When the aganglionosis is restricted to the rectosigmoid colon it is known as short-segment
disease. These represent the vast majority (80%) of cases of HD. However, in approximately 15% to
20% patients the aganglionosis extends proximal to the sigmoid colon which is designated as longsegment
disease. In about 5% cases the aganglionosis involves the entire colon called total colonic
aganglionosis. In extremely rare situation the aganglionosis is seen extending into small intestine
or even more proximally, the so called total intestinal aganglionosis [3]. This condition is invariably
fatal [4].
The patients typically present with inability to pass meconium within first 48 hours of life.
The other modes of presentation include constipation, vomiting along with painful and distended
abdomen. The abdominal distension can be sometimes quite massive due to huge distension of the
colon known as megacolon. However, sometimes it presents in older children or adult. Thus in cases
presenting with chronic constipation in these age groups possibility of HD should be kept in mind.
The diagnosis of HD is established by histopathological examination to show absence of
ganglion cells in submucosal plexus. Suction biopsy is the preferred method as it is safe and does
not require general anesthesia. Other findings seen in biopsies include hypertrophic nerve fibers
[5]. Acetylcholinesterase histochemistry shows abnormally increased nerve fibers in the mucosa [6]. Other supporting investigations include anorectal manometry,
abdominal radiographs with barium enema. Intraoperative frozen
sections help in planning an accurate surgery.
The phenotype of these patients is however highly variable. This
can be explained on the basis of the genetic abnormalities and the
interactions between the various genes in cases of HD. An overview
of the current understanding of this highly complex genetic landscape
is presented in this article.
Table 1
Table 1
Chromosomal abnormalities seen in association with HD and their underlying gene loci.
Table 2
Table 2
Syndromes associated with HD.
Genetic Abnormalities in Hirschprung Disease
The genetic abnormalities can be broadly classified into
chromosomal anomalies and single gene abnormalities.
Chromosomal anomalies
The overall frequency of chromosomal aberrations in HD is about
12% [7]. The strongest association of HD is with Down syndrome
(Trisomy 21) which has been reported to vary from as low as 2% to
as high as 10% [8].
It is however interesting to note that chromosome 21 does not
contain any gene which is known to be associated with HD. Therefore,
in addition to trisomy 21 there are other chromosomal abnormalities
which specifically involve HD associated genes like EDNRB, RET,
NRG3, ZEB2 and PHOX2B. In case of all of these genes both the alleles
need to be functional for normal status. In case even a single allele
gets inactivated due to mutation there will be clinical manifestation.
This loss of single allele is known as haploinsufficiency. The details of
the chromosomal abnormalities leading to HD are presented in Table
1. It was observed that only some of the patients of HD possessed
some of these chromosomal anomalies that are every patient of HD
does not carry all of these anomalies. On further investigation the
causative genes were also identified. However, as it is quite evident
from Table 1 that still there are chromosomal abnormalities in which
the underlying genes have not been identified.
Single gene mutations
Besides the chromosomal abnormalities as described above a
certain group of HD patients show mutations involving single gene.
The various modes of inheritance that have been reported in these
cases include autosomal dominant, autosomal recessive, or X-linked.
Further these cases may or may not show coexisting syndromes.
HD associated with syndromes
There are large numbers of syndromes that may have an
association with HD. These cases usually present clinically as long
segment HD. The details of these syndromes are summarily presented
in Table 2.
HD not associated with syndromes
In these cases of HD there is no definite association with any
congenital anomaly. Broadly describing these cases show mutation in
either one of these four genetic pathways
1. RET along with its ligands GDNF and NRTN
2. EDNRB and its associated genes EDN3 and ECE1
3. NRG pathway and the genes involved being NRG1 and
NRG3
4. SEMA signaling pathway and the related genes SEMA3C
and SEMA3D
The details of the genes involved in these cases of HD are shown
in Table 3.
HD associated with congenital abnormalities of unknown
etiology
There are certain cases of HD which have at least one congenital
abnormality associated with them. However, it is not possible to
place these cases into definite syndromes. The currently available
information about these cases of HD is summarized in Table 4.
Figure 1
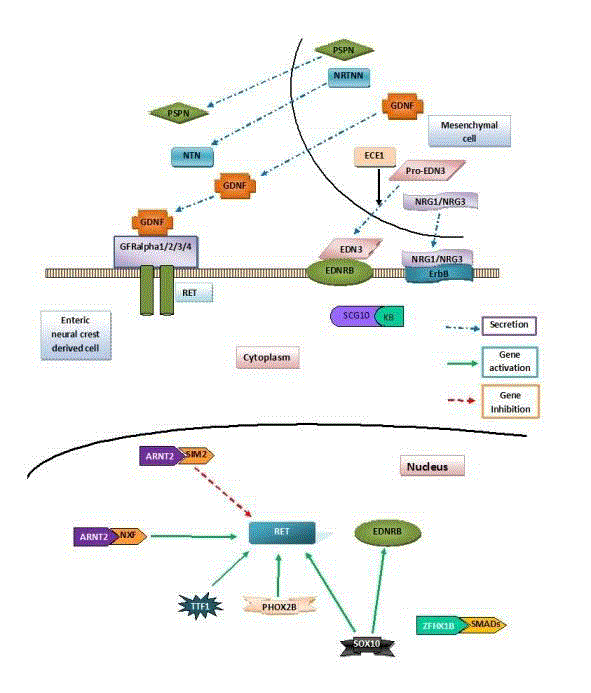
Figure 1
Role of RET, EDNRB and NRG pathways in HD: Giant cell
derived neurotrophic factor (GDNF) secreted by the mesenchymal cells
(Blue dashed arrow) binds to its receptor RET on the enteric neural crest
derived cell. This causes proliferation of these cells by further downstream
mechanisms. Endothelin-3 (EDN3) also secreted by the mesenchymal
cells goes and binds to its receptor Endothelin receptor B (EDNRB) on the
enteric neural crest derived cells. This causes their proliferation and survival.
PHOX2B and SOX-10 increase the expression (green solid arrow) of RET
and EDNRB genes. SIM2 inhibits the expression of RET (red dashed arrow).
Figure 2
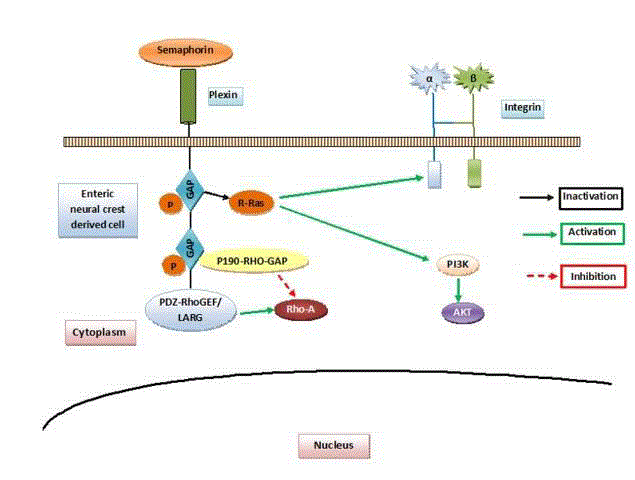
Figure 2
Role of semaphorins: Semaphorins bind to the Plexin family
receptors. Plexins in turn can cause activation of Plexin associate tyrosine
kinases. These plexins have intrinsic GTPase activating protein (GAP)
activity which in turn leads to inactivation of R-Ras (black solid arrow). This
leads to inhibition of integrin mediated activities and PI3K mediated AKT
pathway. Another cytoplasmic domain of Plexins regulates the activation of a
small GTPaseRhoA (red dashed arrow).
Table 3
Table 3
Genes involved in cases of HD that are not associated with any syndromes.
Table 4
Table 4
HD associated with congenital anomalies of an unknown etiology.
Approaches Taken to Study the Genetic Landscape of Hirschprung Disease
Genome wide association studies (GWAS)
Genome wide association studies are those studies in which the
DNA sample is analyzed for the presence of various single nucleotide
pleomorphisms (SNPs) in the genome. Those SNPs which are found
to be more frequent in patients are said to be associated with that
particular disease. Thus to find additional loci that contribute to
the development of HD, a GWAS was carried out in Chinese cases
with sporadic HD [30]. As expected, most frequent association was
found with the RET gene. However, two additional SNPs located in
intron 1 of Neuregulin-1 gene (NRG1) were also found to be strongly
associated, pointing to NRG1 as a plausible candidate gene. This
was further corroborated by the identification of coding sequence
mutations in NRG1 [30-32]. Copy number variation analysis on this
data revealed another gene to be associated with HD, namely NRG3,
a paralog of NRGI. Further validation on Chinese HD patients
identified nine deletions and two de nono duplications in NRG3,
suggesting a role of this gene in HD [33].
A novel pathway based analysis approach was used by Fernandez
et al. [34] to initially prioritize candidate genes in a Spanish cohort
of 53 cases of short-segment Hirschsprung disease. Candidate genes
were further validated in an independent population of 106 cases.
Their study revealed a strong association of 11 Gene Ontology (GO)
modules which were related to signal transduction and its regulation,
Enteric Nervous System (ENS) formation and other HD-related
processes. Among the preselected genes, a total of 4 loci, RASGEF1A,
IQGAP2, DLC1 and CHRNA7, related to signal transduction and In order to discover additional genetic loci, Kim et al. [35]
performed a GWAS of 123 sporadic HD patients and 432 unaffected
controls using a large-scale platform. They also found mutation in
the RET-CSGALNACT2-RASGEF1A genomic region and NRG1 as
susceptibility loci. In addition, they identified SLC6A20 and ABCC9
as new potential susceptibility loci. Although none of the SNPs in
these genes passed the Bonferroni correction. In further subgroup
analysis it was observed that the RET-CSGALNACT2-RASGEF1A
genomic region had a differential significance pattern amongst
the various types of HD. This suggests that other genomic loci or
mechanisms may affect the length of aganglionosis in HD subgroups
during Enteric Nervous System (ENS) development.
Another potentially significant locus SLC6A20 was studied by
Lee and coworkers [36]. SLC6A20 stands for solute carrier family
6, proline IMINO transporter, member 20 (SLC6A20). A total
of 40 single nucleotide polymorphisms (SNPs) of SLC6A20 were
genotyped in 187 HD subjects composed of 121 short-segment HD,
45 long-segment HD, 21 total colonic aganglionosis and 283 controls.
The data analysis revealed that 13 SLC6A20 SNPs were significantly
associated with HD. In further subgroup analysis, SLC6A20
polymorphisms appeared to have increased association with L-HSCR.
Thus the results of their study suggest that SLC6A20 may have a role
in aetiopathogenesis of HD and it may also contribute in determining
the length of aganglionic segment.
The probability that a variant (rs6509940) of interleukin-11 (IL-
11) may act as a potential locus for HD was studied by Kim et al. [37].
They further studied associations with HD of nine common SNPs
on IL-11. A total of nine SNPs on IL-11 were genotyped in 187 HD
patients and 283 controls using TaqMan genotyping assay. The data
analysis revealed that several SNPs showed a statistically significant
association with HD. Moreover, the most common haplotype was
strongly associated with HD. In further analysis among three HD
subtypes (short segment, long segment, total colonic aganglionosis),
the results showed a different association pattern depending on
the subtype. This suggests that genetic variations of IL-11 may be
associated with the risk of HD.
Bae et al. [38] attempted to identify new HD genetic factors related
to Copy Number Variation (CNV) and loss of heterozygosity (LOH)
in Korean patients. They performed genome-wide genotyping, using
Illumina's HumanOmni1-Quad BeadChip (1,140,419 markers), on
123 HD patients and 432 controls. A total of 8,188 CNVs (1 kb 1 mb)
were identified by CNV partition. As a result, 16 CNV regions and 13
LOH regions were identified as associated with HD. Two top CNV
regions (deletions at chr6:32675155-32680480 and chr22:20733495-
21607293) were successfully validated by additional real-time
quantitative polymerase chain reaction analysis. In addition, 2 CNV
regions (6p21.32 and 22q11.21) and 2 LOH regions (3p22.2 and
14q23.3) were discovered to be unique to the HD patients group.
Large-scale chromosomal aberrations (>1 mb) were identified in 11
HD patients.
A trans-ethnic meta-analysis of 507 HD cases and 1191 controls
was carried out by Tang et al. [38]. They combined all published
GWAS results on HD. It was seen that the effects of RET and NRG1
are universal across European and Asian ancestries. In contrast, a
European-specific association was observed with a low-frequency
variant, rs80227144, in SEMA3 gene. Further conditional analyses
on the lead SNPs showed a secondary association signal, which
corresponded to an Asian-specific, low-frequency missense variant
encoding RET p.Asp489Asn (rs9282834). When in trans with the
RET intron 1 enhancer risk allele, rs9282834 increases the risk of HD
from 1.1 to 26.7. This is the largest meta-analysis study on HD and
provides great insights into the genetic architecture of HD.
Next generation sequencing
A major advance in molecular biology in the last decade has been
the availability of next generation sequencing as an investigative tool.
While the speed has increased manifold the cost has dramatically
come down. In HD this tool has been applied by some authors leading
to discovery of newer genetic mutations.
In order to evolve an efficient approach for identifying rare
mutations which could be possibly related to Hirschsprung disease
(HSCR), Gui et al. [40] carried out a pilot study using a newly developed
protocol for next generation targeted resequencing. A total of 20 HD
patients and 20 sex-matched individuals with no HD as controls
were included. In these patients coding sequences (CDS) of 62 genes
known to be involved in signaling pathways related to enteric nervous
system development were selected for capture and sequencing. The
blood samples of these 40 cases were pooled to make a total of 8 pools.
Each pool comprising of 5 patients. The pooled DNAs was enriched
by PCR¬based Rain Dance technology (RDT) and then sequenced
on a 454 FLX platform. For technical validation, five patients from
Pool-3 were also independently enriched by RDT, indexed with
barcode and sequenced with sufficient coverage. The evaluation of
single nucleotide variants showed that DNA pooling performed well
(specificity/sensitivity at 98.4%/83.7%) for the common variants. But
in case of rare variants it did relatively worse (specificity/sensitivity
at 65.5%/61.3%). Sanger sequencing only validated five out of 12 rare mutations which were reported. Thus the authors suggest that more
technical improvement is required in sequencing technology for
variant detection for large-scale resequencing of pooled DNA.
In a subsequent study Luzón-Toro and coworkers carried out a
study with the aim of designing of a panel of HD associated genes
which could be used to carry out genetic screening. They performed
NGS-based targeted sequencing (454-GS Junior) using a panel
containing 26 associated or candidate genes in 11 patients of HD.
The most notable finding in their study was that they found a total
of 13 new coding variants and 11 new variants within the regulatory
regions.
In another study Luzón-Toro et al. [42] performed whole exome
sequencing in familial HD cases (n=16). They used the SOLID platform
for their study. They looked for genes that were recurrently mutated.
They found that variations in the FAT3 gene were significantly
seen. Within-family analysis showed compound heterozygotes for
ANNAK and several genes (n=23) with heterozygous variants that
co-segregated with the phenotype. Network and pathway analyses
led to the discovery of polygenic inheritance involving FAT3, HD
associated genes and their gene partners. Their approach led to the
detection of more than one damaging variants in genes that could
together contribute to the overall phenotype. They concluded that
these findings further elucidate the complex interactions that occur
during enteric nervous system development and the etiopathogenesis
of familial HD.
Understanding the genetic pathways
The important genes involved in HD have been mentioned above
and shown in Table 3. Based on current understanding majority of
these genes involved in HD can be divided into four groups based on
the pathways in which they are involved. These are: RET activation
pathway (RET, GDNF, PSPN), EDNRB pathway (EDNRB, EDN3,
ECE1), transcription factors involved in both pathways (SOX 10,
PHOX2B) and semaphorin pathway (SEMA3C, SEMA3D). Out of
these RET and EDNRB pathways are relatively well known. As shown
in Figure 1 giant cell derived neurotrophic factor (GDNF) which is
secreted by the mesenchymal cells binds to its receptor RET on the
enteric neural crest derived cell. This causes proliferation of these
cells by further downstream mechanisms. Endothelin-3 (EDN3)
also secreted by the mesenchymal cells goes and binds to its receptor
Endothelin receptor B (EDNRB) on the enteric neural crest derived
cells. This causes their proliferation and survival.
Semaphorin pathway is however a more recently discovered
pathway. Semaphorins are proteins which were originally discovered
for their role in assembly of neural circuitry [43,44]. Their role in HD
has been investigated by several authors. Wang et al. [45] examined
expression of semaphorin 3A in different colonic segments of HD
patients. They studied expression levels of SEMA3A in both ganglionic
and aganglionic colon tissues of 32 patients with HD and in colon
tissue of 5 normal newborns. The tissues were examined by realtime
RT-PCR, Western-blot, and immune histology. Comparison
of SEMA3A expression levels between ganglionic and aganglionic
tissues in HD revealed up regulation of SEMA3A expression in
43.75% (14/32) of the aganglionic colons. SEMA3A was expressed in
the ganglion cells of the myenteric plexus, submucosa, as well as in
the longitudinal and circular muscle layer of the normal colon of both
unaffected newborns and patients with HSCR. In the aganglionic
segment of patients with HD, SEMA3A was highly expressed in
the circular muscle layer and was also detected in the submucosa
and in the longitudinal muscles layer. The fluorescence intensity of
SEMA3A in the circular muscle layer in the aganglionic segment was
much higher than that in ganglionic segment. Thus they concluded
that SEMA3A expression was upregulated in the aganglionic smooth
muscle layer of the colon in some patients with HD and increased
SEMA3A expression may be a risk factor for HD pathology in a
subset of patients.
In another subsequent studyLuzón-Toro et al. [46] carried out a
comprehensive analysis of SEMA3A and SEMA3D in a series of 200
Spanish HD patients. RET mutations were also detected in some of
those patients carrying SEMAs mutations. They evaluated the A131TSEMA3A,
S598G-SEMA3A and E198K-SEMA3D mutations using
colon tissue sections by immunohistochemistry. All mutants showed
increased protein expression in smooth muscle layer of ganglionic
segments. Moreover, A131T-SEMA3A also maintained higher
protein levels in the aganglionic muscle layers. These findings strongly
suggest that these mutants have a pathogenic effect on the disease.
Furthermore, coexistence with RET mutations, further substantiates
the additive genetic model proposed for this rare disorder and further
support the association of SEMAs genes with HD.
The major receptors for semaphorins are Plexin family receptors.
Plexins in turn can cause activation of Plexin associate tyrosine
kinases. Besides this plexins have intrinsic GAP activity which in
turn leads to inactivation of an important G-protein R-Ras. This
leads to inhibition of integrin mediated cell adhesion and the rest
of downstream events. Another cytoplasmic domain of Plexins via
signal transducers regulates the activation of a small GTPaseRhoA. In
addition, GTPases are known to play important role in cell motility.
Thus it is plausible that semaphorin mediated inhibition of cell
motility may be responsible for migration failure of enteric neural
crest derived cells in some cases of HD (Figure 2).
Role of genetic counseling
Since presently the understanding of the genetics of HD is not
complete the role of genetic counseling is not clear. But it is likely that
testing methodology and our understanding of genes, allelic variants, and diseases will improve in the future. Thus, it is expected that genetic
counseling will likely play an increasingly more important role in the
management of these cases. DNA banking of affected patients may
also be of value in the future.
Prenatal testing and preimplantation genetic diagnosis
Prenatal testing and preimplantation genetic diagnosis are
presently not routinely performed in the cases of HD. However,
since many of the pathogenic genetic variants are now well known
this testing modality may be offered to some of the cases especially
syndromic HD. However, one thing must be kept in mind that
presence of a mutation does not necessarily mean that the child will
develop clinical manifestations of HD. This fact must be discussed
with the parents and they must be allowed to make an informed
choice.
Conclusion
Thus to conclude HD is an extremely complex disease at genetic level. There is much more to be studied and learned before we are able to fully unravel this disease. The task at hand is proving to be extremely difficult. This is because a combinations of mutations together lead to the disease. Some of these are common and others are rare. But what is proving quite difficult is to associate a particular mutation to the disease. The present approach is to use NGS and GWAS. For any new variant discovered in these studies it is important to do functional analysis studies to prove their role in the pathogenesis of HD. In the future it is believed that a combination of these approaches and the development of an animal model will lead to further improvement in understanding of this disease.
References
- Parisi MA, Kapur RP. Genetics of Hirschsprung disease. Curr Opin Pediatr. 2000;12(6):610-7.
- Heanue TA, Pachnis V. Enteric nervous system development and Hirschsprung's disease: advances in genetic and stem cell studies. Nat Rev Neurosci. 2007;8(6):466-79.
- Badner JA, Sieber WK, Garver KL, Chakravarti A. A genetic study of Hirschsprung disease. Am J Hum Genet. 1990;46(3):568-80.
- Ruttenstock E, Puri P. A meta-analysis of clinical outcome in patients with total intestinal aganglionosis. Pediatr Surg Int. 2009;25(10):833-9.
- Kapur RP. Hirschsprung disease and other enteric dysganglionoses. Crit Rev Clin Lab Sci. 1999;36(3):225-73.
- Kobayashi H, Miyahara K, Kusafuka J, Yamataka A, Lane GJ, Sueyoshi N, et al. A new rapid acetylcholinesterase staining kit for diagnosing Hirschsprung's disease. Pediatr Surg Int. 2007;23(5):505-8.
- Amiel J, Lyonnet S. Hirschsprung disease, associated syndromes, and genetics: a review. J Med Genet. 2001;38(11):729-39.
- Moore SW, Johnson AG. Hirschsprung's disease: genetic and functional associations of Down's and Waardenburg syndromes. Semin Pediatr Surg. 1998;7(3):156-61.
- Amiel J, Espinosa-Parrilla, Steffann J, Gosset P, Pelet A, Prieur M, et al. Large-scale deletions and SMADIP1 truncating mutations in syndromic Hirschsprung disease with involvement of midline structures. Am J Hum Genet. 2001;69:1370-77.
- Mäkitie O, Kaitila I, Rintala R. Hirschsprung disease associated with severe cartilage-hair hypoplasia. J Pediatr. 2001;138(6):929-31.
- Trang H, Dehan M, Beaufils F, Zaccaria I, Amiel J, Gaultier C, et al . The French Congenital Central Hypoventilation Syndrome Registry: general data, phenotype, and genotype. Chest. 2005;127:72-9.
- Slaugenhaupt SA, Blumenfeld A, Gill SP, Leyne M, Mull J, Cuajungco MP, et al. Tissue-specific expression of a splicing mutation in the IKBKAP gene causes familial dysautonomia. Am J Hum Genet. 2001;68(3):598-605.
- Slavotinek AM. Fryns syndrome: a review of the phenotype and diagnostic guidelines. Am J Med Genet A. 2004;124A(4):427-33.
- Drevillon L, Megarbane A, Demeer B, Matar C, Benit P, Briand-Suleau A, et al. KBP-cytoskeleton interactions underlie developmental anomalies in Goldberg-Shprintzen syndrome. Hum Mol Genet. 2013;22:2387-99.
- Nakakimura S, Sasaki F, Okada, Arisue A, Cho K, Yoshino M, et al. Hirschsprung s disease, acrocallosal syndrome, and congenital hydrocephalus: report of 2 patients and literature review. J Pediatr Surg. 2008;43:E13-7.
- Hansford JR, Mulligan LM. Multiple endocrine neoplasia type 2 and RET: from neoplasia to neurogenesis. J Med Genet. 2000;37(11):817-27.
- Romeo G, Ceccherini I, Celli J, Priolo M, Betsos N, Bonardi G, et al. Association of multiple endocrine neoplasia type 2 and Hirschsprung disease. J Intern Med. 1998;243(6):515-20.
- Mowat DR, Wilson MJ, Goossens M. Mowat-Wilson syndrome. J Med Genet. 2003;40(5):305-10.
- Inoue K, Khajavi M, Ohyama T, Hirabayashi S, Wilson J, Reggin JD, et al. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat Genet. 2004;36(4):361-9.
- Bahuau M, Pelet A, Vidaud D, Lamireau T, LeBail B, Munnich A, et al. GDNF as a candidate modifier in a type 1 neurofibromatosis (NF1) enteric phenotype. J Med Genet. 2001;38(9):638-43.
- Peippo MM, Simola KO, Valanne LK, Larsen AT, Kähkönen M, Auranen MP, et al. Pitt-Hopkins syndrome in two patients and further definition of the phenotype. Clin Dysmorphol. 2006;15(2):47-54.
- Mueller C, Patel S, Irons M, Antshel K, Salen G, Tint GS, et al. Normal cognition and behavior in a Smith-Lemli-Opitz syndrome patient who presented with Hirschsprung disease. Am J Med Genet A. 2003;123A(1):100-6.
- de Pontual L, Pelet A, Trochet D, Jaubert F, Espinosa-Parrilla Y, Munnich A, et al. Mutations of the RET gene in isolated and syndromic Hirschsprung's disease in human disclose major and modifier alleles at a single locus. J Med Genet. 2006;43(5):419-23.
- Angrist M, Bolk S, Halushka M, Lapchak PA, Chakravarti A. Germline mutations in glial cell line-derived neurotrophic factor (GDNF) and RET in a Hirschsprung disease patient. Nat Genet. 1996;14:341-4.
- Sánchez-Mejías A, Fernández RM, López-Alonso M, Antiñolo G, Borrego S. New roles of EDNRB and EDN3 in the pathogenesis of Hirschsprung disease. Genet Med. 2010;12(1):39-43.
- Tang CS, Tang WK, So MT, Miao XP, Leung BM, Yip BH, et al. Fine mapping of the NRG1 Hirschsprung's disease locus. PLoS One. 2011;6(1):e16181.
- Yang J, Duan S, Zhong R, Yin J, Pu J, Ke J, et al. Exome sequencing identified NRG3 as a novel susceptible gene of Hirschsprung's disease in a Chinese population. Mol Neurobiol. 2013;47(3):957-66.
- Jiang Q, Arnold S, Heanue T, Kilambi KP, Doan B, Kapoor A, et al. Functional loss of semaphorin 3C and/or semaphorin 3D and their epistatic interaction with ret are critical to Hirschsprung disease liability. Am J Hum Genet. 2015;96(4):581-96.
- Gunadi, Makhmudi A, Agustriani N, Rochadi. Effects of SEMA3 polymorphisms in Hirschsprung disease patients. Pediatr Surg Int. 2016;32(11):1025-1028.
- Garcia-Barcelo MM, Tang CS, Ngan ES, Lui VC, Chen Y, So MT, et al. Genome-wide association study identifies NRG1 as a susceptibility locus for Hirschsprung's disease. Proc Natl Acad Sci U S A. 2009;106(8):2694-9.
- Luzón-Toro B, Torroglosa A, Núñez-Torres R, Enguix-Riego MV, Fernández RM, de Agustín JC, et al. Comprehensive analysis of NRG1 common and rare variants in Hirschsprung patients. PLoS One. 2012;7(5):e36524.
- Tang CS, Ngan ES, Tang WK, So MT, Cheng G, Miao XP, et al. Mutations in the NRG1 gene are associated with Hirschsprung disease. Hum Genet. 2012;131(1):67-76.
- Tang CS, Cheng G, So MT, Yip BH, Miao XP, Wong EH, et al. Genome-wide copy number analysis uncovers a new HSCR gene: NRG3. PLoS Genet. 2012;8(5):e1002687.
- Fernández RM, Bleda M, Núñez-Torres R, Medina I, Luzón-Toro B, García-Alonso L, et al. Four new loci associations discovered by pathway-based and network analyses of the genome-wide variability profile of Hirschsprung's disease. Orphanet J Rare Dis. 2012;7:103.
- Kim JH, Cheong HS, Sul JH, Seo JM, Kim DY, Oh JT, et al. A genome-wide association study identifies potential susceptibility loci for Hirschsprung disease. PLoS One. 2014;9(10):e110292.
- Lee JS, Oh JT, Kim JH, Seo JM, Kim DY, Park KW, et al. Association Analysis of SLC6A20 Polymorphisms With Hirschsprung Disease. J Pediatr Gastroenterol Nutr. 2016;62(1):64-70.
- Kim LH, Cheong HS, Shin JG, Seo JM, Kim DY, Oh JT, et al. Genetic variants of IL-11 associated with risk of Hirschsprung disease. Neurogastroenterol Motil. 2015;27(10):1371-7.
- Bae JS, Koh I, Cheong HS, Seo JM, Kim DY, Oh JT, et al. A genome-wide association analysis of chromosomal aberrations and Hirschsprung disease. Transl Res. 2016;177:31-40.
- Tang CS, Gui H, Kapoor A, Kim JH, Luzón-Toro B, Pelet A, et al. Trans-ethnic meta-analysis of genome-wide association studies for Hirschsprung disease. Hum Mol Genet. 2016;25(23):5265-75.
- Gui H, Schriemer D, Cheng WW, Chauhan RK, Antiňolo G, Berrios C, et al. Whole exome sequencing coupled with unbiased functional analysis reveals new Hirschsprung disease genes. Genome Biol. 2017;18(1):48.
- Luzón-Toro B, Espino-Paisán L, Fernández RM, Martín-Sánchez M, Antiñolo G, Borrego S. et al. Next-generation-based targeted sequencing as an efficient tool for the study of the genetic background in Hirschsprung patients. BMC Med Genet. 2015;16:89.
- Luzón-Toro B, Gui H, Ruiz-Ferrer, Sze-Man Tang C, Fernández RM, Sham PC M, et al. Exome sequencing reveals a high genetic heterogeneity on familial Hirschsprung disease. Sci Rep. 2015;5:16473.
- Kolodkin AL, Matthes DJ, Goodman CS. The semaphorin genes encode a family of transmembrane and secreted growth cone guidance molecules. Cell. 1993;75(7):1389-99.
- Luo Y, Raible, Raper JA. Collapsin: a protein in brain that induces the collapse and paralysis of neuronal growth cones. Cell. 1993;75(2):217-27.
- Wang LL, Fan Y, Zhou FH, Li H, Zhang Y, Miao JN, et al. Semaphorin 3A expression in the colon of Hirschsprung disease. Birth Defects Res A Clin Mol Teratol. 2011;91(9):842-7.
- Luzón-Toro B, Fernández RM, Torroglosa A, de Agustín JC, Méndez-Vidal C, Segura DI, et al. Mutational spectrum of semaphorin 3A and semaphorin 3D genes in Spanish Hirschsprung patients. PLoS One. 2013;8(1):e54800.