Case Report
A Case of a Typical Juvenile Hyaline Fibromatosis
Karthik Vishwanath, Nikhil S Shetty and Abhishek A Jha*
Department of Plastic Surgery, KS Hegde Medical Academy, India
*Corresponding author: Abhishek A Jha, Department of Plastic Surgery, K.S.Hegde Medical Academy, Mangalore, karnataka, India
Published: 06 Mar, 2017
Cite this article as: Vishwanath K, Shetty NS, Jha AA.
A Case of a Typical Juvenile Hyaline
Fibromatosis. Clin Surg. 2017; 2: 1345.
Abstract
Juvenile hyaline fibromatosis (JHF) is a rare, autosomal recessive disease characterized by papular and nodular skin lesions, gingival hyperplasia, joint contractures and bone involvement in variable degrees. It typically becomes apparent at birth or in infancy, causing severe pain with movement and progressive joint contractures. It is caused by mutations in the ANTXR2 gene. Histopathological examination (HPE) of the cutaneous lesions is unique and characterized by an accumulation of an amorphous, hyaline material in the dermis with increased number of fibroblasts. This disease has a progressive course with most patients surviving only up to the 4 decade. As of now, there is no specific treatment for this disorder. The treatment is only aesthetic and its aim is to limit orthopedic disability. We herein present a case report of 40 year old male with history of multiple swellings over the body since age 1. He has history of multiple surgeries for excision of swelling with recurrence reported at operated site. It is worthwhile considering that as per literature only 70 cases has been reported worldwide.
Introduction
Juvenile hyaline fibromatosis is a rare, autosomal recessive disease characterized by popular and nodular skin lesions, gingival hyperplasia, joint contractures and bone involvement in variable degrees [1]. It typically becomes apparent at birth or in infancy, causing severe pain with movement and progressive joint contractures [1]. Other features may include digestive problems, gum enlargement; skin bumps; pearly papules on the face and neck; and perianal masses. Complications can be life threatening. It is caused by mutations in the ANTXR2 gene [1,2]. We herein present a case report of a young man in his 30s with history of multiple swellings over the body since age 1. He has history of multiple surgeries for excision of swelling with recurrence reported at operated site. It is worthwhile considering that as per literature only 70 cases has been reported worldwide [6].
Case Presentation
A young gentleman presented with history of multiple swellings that begun at the age 1. His family noticed one swelling at the back of head for which they underwent excision. Following the surgery patient developed a larger swelling at the operated site and multiple swellings over the whole body (Figure 1-3). He underwent surgeries to remove swellings for almost 13 times. He has no other complaints or any co-morbidity; no significant family history; no problems in speaking or eating or in movements of any other joints. His clinical examination has been normal (Figure 4). Multiple swellings were noted on his scalp back, hands and feet, waist. The swellings were cystic in consistency, restricted mobility (Figure 5). We noticed a few osteolytic lesions on X ray of the upper and lower limbs while the other x rays were normal (Figure 6). After a thorough clinical and radiological work up he was taken up for tumorectomy with a differential of neurofibromatosis in mind (Figure 7). Complete excision of tumour was not possible hence debulking was performed. Intra operatively we found white fibrous material in the tumour bed that bled continuously (Figure 8). The specimen was sent for histopathological examination and it turned out to be a HYALINE FIBROMATOSIS
Figure 1-3
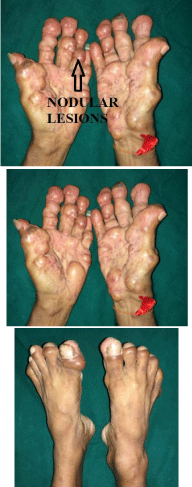
Figure 1-3
Nodular lessions.
Figure 4
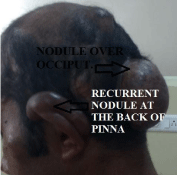
Figure 4
Recurrent nodule at the back of pinna.
Figure 5
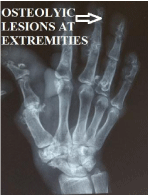
Figure 5
Osteolyic lesions at extremities.
Figure 6
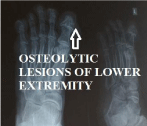
Figure 6
Right sided lumber nodule for excision.
Figure 7
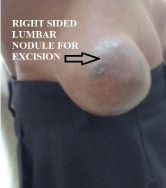
Figure 7
Benign oval to spindle shaped cells in cords.
Figure 8
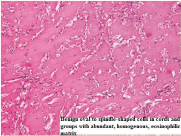
Figure 8
Osteolytic lesionss of lower extremity.
Discussion
Juvenile hyaline fibromatosis (JHF) was originally described by Murray in 1873 under the name “molluscum fibrosum”. At that time, it was considered a variant of neurofibromatosis. Whitfield and Robinson later reported two more cases in 1903 and suggested the disease be recategorized as multiple fibromata, but it was not until 1972 when this condition was given the current name of juvenile hyaline fibromatosis by Kitano et al [1]. An autosomally recessive mode of inheritance is accepted, but sporadic cases can occur. The gene that causes JHF has been mapped to 4q21 and mutations in the capillary morphogenesis have also been described [1]. The exact pathogenesis is unknown but several theories have been proposed, most attributing JHF lesions either to aberrant synthesis of glycosaminoglycans by fibroblasts or to disordered collagen metabolism [1,2]. JHF is a rare hereditary disease with progressive course that should be highly suspected in a patient with early onset papulonodules, joint contractures and gingival hypertrophy. It usually affects children less than 5 years of age [3]. Joint contracture and gingival hypertrophy precede the skin manifestation. Early diagnosis and proper management are crucial in an attempt to slow the progression of this rare disabling disease. Musculoskeletal involvement in JHF is frequent, and flexion contracture of large joints is the most debilitating problem; most adolescents and adults become bedridden and die of infection [4]. It has been hypothesized that contractures result from infiltration of the capsules of the joints. Gilaberte proposed 2 major and 3 minor diagnostic criteria for JHF. Major criteria include cutaneous lesions and gingival enlargement. The skin lesions may present as small pearly papules (generally on the face and neck), small nodules, and large plaques with translucent appearance and gelatinous consistency (on fingers, ears, and around the nose) or large, firm subcutaneous tumors (most common on the scalp, trunk, and limbs). Minor criteria include joint contractures, osteolytic bone lesions and/or cortical erosion, and a family history of JHF [5]. The differential diagnosis of JHF includes neurofibromatosis, gingival fibromatosis, nodular amyloidosis, infantile congenital generalized fibromatosis, lipoid proteinosis and Winchester syndrome. The present case has only cutaneous lesions, but there is no gingival hypertrophy, joint or bone involvement till date. The tumor masses are variable in size, slow growing and painless and have a tendency to recur following excision. The diagnosis of JHF can be confirmed by histology. The tumors are poorly circumscribed and consist of cords of spindleshaped cells embedded in a homogeneous eosinophilic matrix. They are often found in the dermis, subcutis and gingiva, although the bone and joints may also be involved. Quintal and Jackson reported a patient who had numerous surgical excisions over a period of 34 years and found that the therapy was as mutilating as the disease [6]. This disease has a progressive course with most patients surviving only up to the 4 decade. As of now, there is no specific treatment for this disorder. The treatment is only aesthetic and its aim is to limit orthopedic disability. Early tumorectomy may help, but relapses are common. Genetic counselling helps to explain to parents about a 25% chance of having a diseased baby in any pregnancy.
Conclusion
It is worth considering diagnosis of hyaline fibromatosis as a differential in patients presenting with multiple swellings over the body. Identification of the gene mutation responsible for this condition will allow insight into the fundamental defect that causes this rare disorder and provide better treatment for patients with this disease. With the gene for the disease being mapped recently, techniques or antenatal diagnosis are likely to be established.
References
- Mohammed H Abduljabbar. A case report of juvenile hyaline fibromatosis. Journal of Dermatology & Dermatologic Surgery. 2014; 18: 38-42.
- Jayashree Krishnamurthy, Bibhas Saha, Manjunath Gubanna. Juvenille Hyaline Fibromatosis. Indian J Dermatol. 2011; 56: 731-733.
- Quintal D, Jackson R. Juvenile hyaline fibromatosis: A 15year follow-up. Arch Dermatol. 1985; 121: 1062-1063.
- Gilaberte Y, Gonzalez-Mediero I, Lopez Barrantes V, Zambrano A. Juvenile hyaline fibromatosis with skull-encephalic anomalies: A case report and review of the literature. Dermatology. 1993; 187: 144-148.
- Nischal KC, Sachdev D, Kharkar V, Mahajan S. Juvenile hyaline fibromatosis. J Postgrad Med. 2004; 50: 125-126.
- Gupta LK, Singhi MK, Bansal M, Khullar R, Jain V, Kachhawa D. Juvenile Hyaline Fibromatosis in siblings. Indian J Dermatol Venereol Leprol. 2005; 71: 115-118.