Case Report
Sustained Efficacy of Targeted Therapy with Vemurafenib in BRAF(V600e) - Mutated Erdheim - Chester Disease
Kimberly D. Tran, Juan Ayala-Haedo, Oliver Fischer, Sander R. Dubovy and Sara T. Wester*
Department of Ophthalmology, University of Miami, USA
*Corresponding author: Sara T. Wester, Department of Ophthalmology, Bascom Palmer Eye Institute, University of Miami, 900 NW 17th St. Miami, FL 33136, USA
Published: 06 Dec, 2016
Cite this article as: Tran KD, Ayala-Haedo J, Fischer O,
Dubovy SR, Wester ST. Sustained
Efficacy of Targeted Therapy with
Vemurafenib in BRAF(V600e) - Mutated
Erdheim - Chester Disease. Clin Surg.
2016; 1: 1230.
Abstract
The authors report the case of a 45-year-old African American female with complex history of
pituitary mass, diabetes insipidus, anasarca, congestive heart failure, right atrial mass, cardiac
tamponade, pericardial effusions, retroperitoneal fibrosis, and lower extremity arthralgia, who
presented for evaluation of bilateral exophthalmos and intraconal orbital masses. Orbital biopsy
revealed histiocytic infiltrate with immunohistochemical staining positively for CD 68, and
negatively for S100 and CD1a, as well as genetic testing positive for the proto-oncogene B-Raf
protein (BRAFV600E) mutation. The presentation was suggestive of Erdheim-Chester Disease, and
the patient was treated with Vemurafenib with significant sustained clinical improvement.
Keywords: Erheim-chester disease; Xanthogranuloma; Vemurafenib; BRAF(V600E)
Introduction
Erdheim-Chester disease (ECD) is a rare non-Langerhans cell histiocytosis with multi-organ involvement, characterized by tissue infiltration by lipid-laden histiocytes. ECD presents a challenge for clinicians as the clinical presentation, course, and prognosis are all highly influenced by site of disease involvement. The severity of ECD course ranges from asymptomatic to life threatening, with a reported global 5-year mortality of 30-40% [1]. We describe a patient that presented to our clinical institution with a history of multi-organ disease and orbital involvement with worsening of disease despite one year of high dose corticosteroids and Methotrexate for systemic inflammatory disease of unknown etiology.
Case Presentation
A 45-year old African American female presented to the oculoplastics service due to a 4
month history of bilateral eye pain, proptosis, and diplopia. Her complex medical history began
3 years prior when she had a seizure episode due to pituitary tumor and diabetes insipidus (DI),
for which she was treated medically. She was readmitted the following year for renal failure due
to retroperitoneal fibrosis associated with hydronephrosis, and required nephrostomy tube and
ureteral stent placement. A diagnosis of histiocytosis was raised at that time; however, biopsy of
the retroperitoneal mass showed only non-specific fibrotic changes, and thus the diagnosis was not
established. The patient was treated for one year with high dose steroids and Methotrexate, and
the following year, despite treatment, she was re-hospitalized for pericardial tamponade due to a
large pericardial effusion, requiring a pericardial window placement. Over the months following,
the patient’s symptoms worsened. She developed marked anasarca and shortness of breath, and
further workup revealed congestive heart failure due to a right atrial mass.
On presentation to the oculoplastics service, the patient was noted to have bilateral severe orbital
pain and proptosis (Figure 1A and 1B). She demonstrated bilateral upper lid fullness, bilateral
lower lid retraction, and limitation to supraduction and adduction in the right eye. The remaining
ophthalmic exam was unremarkable. Best corrected visual acuity was 20/20 in both eyes, and
there was no evidence of optic nerve compromise. MRI imaging revealed bilateral perioptic nerve
sheath masses, displacing but not involving the optic nerves, in a pattern atypical of meningioma
(Figure1C and 1D). There was also enlargement of intraconal and preseptal orbital tissues with
radiographic evidence of lacrimal gland involvement. Due to concern for IgG4-related disease, the
patient underwent a lacrimal gland biopsy which demonstrated non-specific inflammatory changes.
Due to the inconclusive biopsy results as well as persistent concern for IgG4-related disease given
her clinical and radiographic presentation, another biopsy was performed, this time of the medial
intraconal orbital tissues. Histopathologic analysis revealed xanthogranulomatous inflammation
with reactive fibrosis and a moderate amount of histiocytes which
stained positively for CD-68, and negative for S-100 and CD1a
(Figure 2A-2D). These histopathologic findings in the context of
her clinical presentation were suggestive of ECD, and the patient
was referred to Rheumatology and Hematology-Oncology for comanagement.
While initial flow cytometric analysis of peripheral
blood and bone marrow biopsy were unremarkable, the patient
subsequently developed severe pain in both knees to the point of
difficulty ambulating. Gallium scan showed evidence of increased
absorption of the long bone juxtaarticular areas (Figure 3). Due
to reports of the characteristic B-raf proto-oncogene mutation
(BRAFV600E) in cases of ECD, the orbital biopsy sample was sent for
genetic analysis. The biopsy sample was positive for the BRAFV600E
mutation, and the patient who had initially started Interferon-alpha
(IFNα) as initial empiric therapy, was switched to Vemurafenib, with
subsequent improvement in her orbital pain, proptosis, and limited
ductions. MRI at 9 months after induction of Vemurafenib therapy
demonstrated persistent intraconal masses with interval improvement
and more orbital fat visible. After 18 months of Vemurafenib therapy,
no drug resistance has emerged, and her orbital and systemic disease
remains stable without exacerbation or progression.
Figure 1
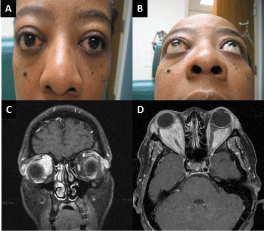
Figure 1
Clinical presentation with (A, B) bilateral proptosis with lower lid retraction and upper eyelid fullness (C) Coronal and (D) Axial T1 Contrast enhanced MRI scans showing bilateral perioptic nerve sheath masses, displacing but not involving the optic nerves, in a pattern atypical of meningioma.
Figure 2
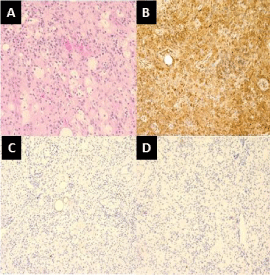
Figure 2
Histopathologic specimen from right medial orbital lesion (40x). (A)
Foamy histiocytes, lymphocytes, and proliferating fibroblasts. These cells are
positive for (B) CD 68 (20x), and negative for (C) S100 and (D) CD1a.
Figure 3
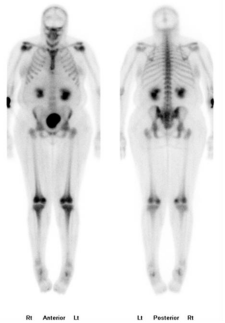
Figure 3
Whole bone scan showing increased uptake on long bones on juxta articular areas.
Discussion
We present the case of a 45-year old female with ECD who was
treated for years for multi-organ disease without a clear diagnosis,
demonstrating the challenge for clinicians in diagnosing and
managing this rare condition. The mean age of presentation is
approximately 55 years, but it can present from adolescence until
adulthood [2]. Men are more frequently affected than women [2].
ECD is included in a spectrum of adult xanthogrogranulomatous
disease which includes Adult onset xanthogranuloma (AOX) (solitary
lesion without systemic findings), Adult onset asthma and periocular
xanthogranuloma (AAPOX), Necrobiotic xanthogranuloma (NBX),
and ECD. While they share similar histopathologic findings of
non-Langerhans histiocytosis, Touton giant cells, varying degrees
of fibrosis, and possible necrosis, they are differentiated by their
systemic disease manifestations, with ECD being associated with the
worst morbidity and mortality.
The disease manifestation of ECD is determined by the organ
system involved, and skeleton, retroperitoneum, and orbit are the
most common sites of involvement, in addition to disease involving
the cardiovascular, pulmonary, neurologic and endocrine systems.
Cardiovascular manifestations include pericardial effusion, sheathing
of the aorta, pericardial effusion, myocardial infarction, valvular heart
disease, and heart failure. Neurologic manifestations include pituitary
lesions associated with diabetes insipidus or other endocrinopathies,
cerebellar or pyramidal syndromes, cognitive impairment, and
cranial nerve palsies. Renal manifestations include retroperitoneal
fibrosis, bilateral hydronephrosis, renovascular hypertension,
ischemic injury and renal failure. Musculoskeletal manifestations
including juxtaarticular involvement with histiocytic infiltration
and sclerosis of long bones and pain in lower limbs [1,3,4]. Orbital
involvement occurs in approximately 25% of cases, and may present
as exophthalmos, retro-orbital pain, oculomotor palsies, or vision
loss [4-5]. Other affected sites include the liver, spleen, thyroid, and
skin [1].
While the organ systems involved may differ in each case of
ECD, the common unifying diagnostic criteria for ECD, as proposed
by Haroche et al. [6] confirmed histologically by tissue biopsy. In
ECD, one finds characteristic lipid-laden histiocytes organized
into polymorphic xanthogranulomas nested among fibrosis, with
CD68-positive and CD1a negative immunostaining. The expression
of markers of the macrophage lineage, such as CD14 and CD68,
confirms the presence of histiocytic cells. In the majority of cases
(80%), markers of dendritic lineage, such as CD1a and S-100 are not
expressed [1]. These immunohistochemical features help distinguish
ECD from Langerhans’ cell histiocytosis, which is a different type
of histiocytosis with multiorgan involvement that may clinically
demonstrate similarities with ECD [7]. A second adjunctive criterion
as proposed by Haroche et al. [6] is the finding of bilateral and
symmetric cortical osteosclerosis of the diaphyseal and metaphyseal
parts of the long bones on X-rays, and/or symmetric and abnormally
intense labeling of the distal ends of the long bones of the legs,
and in some cases arms, as revealed by 99Tc bone scintigraphy. The
second criterion is not strictly required for ECD diagnosis. More
recently, a characteristic histiocyte-restricted mutation in the genetic
sequence of the B-Raf proto-oncogene, which confers the amino
acid substitution of glutamic acid for valine at position 600 of the
B-Raf protein(BRAFV600E), has been associated in at least 50% of the
cases [8-9], lending one additional diagnostic tool in characterizing
this disease. The expression of ECD can range from focal indolent
disease to fulminant multiorgan system dysfunction which in some
cases may be fatal. Thus it is recommended that once the diagnosis is
confirmed, baseline evaluation include computed tomography (CT)
scans of the chest, abdomen, pelvis, positive emission tomography
(PET) of the entire body, magnetic resonance imaging (MRI) of the
brain with gadolinium and cardiac MRI to evaluate the extent of
organ system involvement [3].
The pathogenesis of ECD is not fully understood, and current
data suggest a clonal mechanism in the setting of a local and systemic
pro-inflammatory cytokine-chemokine network which may be
responsible for the recruitment and activation of histiocytes into ECD
lesions [10-11]. BRAF is involved in the regulation of cell proliferation
and survival as it contributes to the RAS-RAF-MEK-ERK protein
kinase pathway. Mutations of BRAF lead to chronic activation of this
pathway leading to cellular proliferation and survival of cells [12-13].
The BRAFV600E mutation, in particular, has oncogenic activity that has
previously been described in melanomas, papillary thyroid cancers
and hairy-cell leukemia [3,8,14,15]. Reports have described >50% of
patients with ECD carrying the BRAFV600E mutation [8,9]. In this setting,
BRAFV600E is thought not only to contribute to clonal proliferation
of BRAFV600E histiocytes but also to contribute to the inflammatory
milieu that is associated with further histiocytic recruitment to ECD
lesions. BRAFV600E has been found to be associated with oncogeneinduced
senescence (OIS), which is a protective mechanism against
oncogenic events. In OIS, an activating mutation in an oncogene
without complementary mutations leads to cell-cycle arrest and
induction of pro-inflammatory molecules. Histiocytes carrying the
BRAFV600E mutation have markers of OIS, thus while the BRAFV600E
mutation promotes clonal proliferation of BRAFV600E histiocytes,
it paradoxically also leads to OIS, and in this way contributes a key
role to the inflammatory milieu observed in recruiting activated
histiocytes to ECD lesions [16]. Activated foamy histiocytes have a
high expression of chemokine/chemokine receptors (CCR) involved
in monocyte migration process (CCR1, CCR2, CCR3, CCR5 and
the ligands CCL4, CCL2, CCL20 and CCL5), and recruited T-helper
lymphocyte demonstrate increased expression of IFNγ and the IFNγ
induced chemokine (IP-10) on histiocytes with an auto feedback loop
that in turn recruit more histiocytes and proinflammatory cells [3].
ECD is characterized by elevated levels of interferon alpha (IFNα),
interleukin (IL)-6, IL-12, and monocyte chemotactic protein-1 as
well as by decreased levels of IL-4 and IL-7 [4 and 10]. Il-6 is involved in
osteoclast differentiation that can lead to osteosclerosis, [4] and IL-6
may also play a role in endothelial cell dysfunction and permeability
seen in pericardial effusions of ECD [17].
There have been no randomized controlled trials in ECD.
Case series have reported, with varying degrees of success, the use
of steroids as well as other immunomodulatory agents. IFNα is
currently considered first-line treatment, which induces activation of
CD40 for immune-mediated destruction of histiocytes. Cladribine is
a purine analogue that causes monocyte and T-lymphocyte depletion
[18]. Anakinra is a recombinant non-glycosylated form of human IL-
1Ra that down regulates the activity of IL-1. Infliximab is a chimeric
monoclonal antibody against TNFα. Recently, Vemurafenib therapy
has been studied and it appears to be a highly promising treatment,
[10] as it induces apoptosis in cell lines expressing the BRAFV600E
mutation. Interestingly, it has also been effective in BRAFV600E mutated
melanoma cell lines [10] and non-melanoma cancer [19]. Given
the patient’s positive BRAFV600E genetic analysis, she was started on
Vemurafenib with significant improvement in her symptoms, which
is consistent with other reports of successful use of Vemurafenib for
ECD [10,20-25]. After 18 months of Vemurafenib therapy, no drug
resistance has emerged, and consistent with other prior reports, her
orbital masses are markedly decreased in size on MRI, [22] and her
systemic disease remains stable without exacerbation or progression.
Conclusion
ECD is a rare disease associated with multidisciplinary morbidity that presently poses a great challenge to the medical care team. The heterogeneity of case presentation due to multi-system involvement makes diagnosis and management particularly challenging, and ECD historically has demonstrated poor response to therapy. More recently, improved understanding into the clonal and pro-inflammatory mechanism of disease and central role of BRAF mutations in this disease may help guide clinicians in the use of anti-inflammatory, immunomodulatory, and anti-proliferative treatments. Long-term outcomes and side effects of these treatments remain important questions, and prospective clinical trials are needed to further guide treatment algorithms for ECD.
References
- Cavalli G, Guglielmi B, Berti A, Campochiaro C, Sabbadini MG, Dagna L, et al. The multifaceted clinical presentations and manifestations of Erdheim-Chester disease: comprehensive review of the literature and of 10 new cases. Ann Rheum Dis. 2013; 72: 1691-1695.
- Arnaud L, Gorochov G, Charlotte F, Lvovschi V, Parizot C, Larsen M, et al. Systemic perturbation of cytokine and chemokine networks in Erdheim- Chester disease: a single-center series of 37 patients. Blood. 2011; 117: 2783-2790.
- Diamond EL, Dagna L, Hyman DM, Cavalli G, Janku F, Estrada-Veras J, et al. Consensus guidelines for the diagnosis and clinical management of Erdheim-Chester disease. Blood. 2014; 124: 483-492.
- Cives M, Simone V, Rizzo FM, Dicuonzo F, Cristallo Lacalamita M, Ingravallo G, et al. Erdheim-Chester disease: a systematic review. Crit Rev Oncol Hematol. 2015; 95: 1-11.
- Haroche J, Arnaud L, Cohen-Aubart F, Hervier B, Charlotte F, Emile JF, et al. Erdheim-Chester disease. Rheum Dis Clin North Am. 2013; 39: 299- 311.
- Haroche J, Arnaud L, Cohen-Aubart F, Hervier B, Charlotte F, Emile JF, et al. Erdheim-Chester disease. Curr Rheumatol Rep. 2014; 16: 412.
- Wilejto M. Langerhans cell histiocytosis and Erdheim-Chester disease. Curr Opin Rheumatol. 2012; 24: 90-96.
- Cangi MG, Biavasco R, Cavalli G, Grassini G, Dal-Cin E, Campochiaro C, et al. BRAFV600E-mutation is invariably present and associated to oncogene-induced senescence in Erdheim-Chester disease. Ann Rheum Dis. 2015; 74: 1596-1602.
- Haroche J, Charlotte F, Arnaud L, von Deimling A, Hélias-Rodzewicz Z, Hervier B, et al. High prevalence of BRAF V600E mutations in Erdheim- Chester disease but not in other non-Langerhans cell histiocytoses. Blood. 2012; 120: 2700-2703.
- Haroche J, Cohen-Aubart F, Emile JF, Arnaud L, Maksud P, Charlotte F, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood. 2013; 121: 1495-1500.
- Campochiaro C. Erdheim-Chester disease. Eur J Intern Med. 2015; 26: 223-229.
- Rahman MT, Nakayama K, Rahman M, Katagiri H, Katagiri A, Ishibashi T, et al. KRAS and MAPK1 gene amplification in type II ovarian carcinomas. Int J Mol Sci. 2013; 14: 13748-13762.
- Emile JF, Diamond EL, Hélias-Rodzewicz Z, Cohen-Aubart F, Charlotte F, Hyman DM, et al. Recurrent RAS and PIK3CA mutations in Erdheim- Chester disease. Blood. 2014; 124: 3016-3019.
- Hyman DM, Diamond EL, Vibat CR, Hassaine L, Poole JC, Patel M, et al. Prospective blinded study of BRAFV600E mutation detection in cell-free DNA of patients with systemic histiocytic disorders. Cancer Discov. 2015; 5: 64-71.
- Mitchell B, Dhingra JK, Mahalingam M. BRAF and Epithelial- Mesenchymal Transition: Lessons From Papillary Thyroid Carcinoma and Primary Cutaneous Melanoma. Adv Anat Pathol. 2016; 244-271.
- Cavalli G, Biavasco R, Borgiani B, Dagna L. Oncogene-induced senescence as a new mechanism of disease: the paradigm of erdheim-chester disease. Front Immunol. 2014; 5: 281.
- Arnaud L, Hervier B, Néel A, Hamidou MA, Kahn JE, Wechsler B, et al. CNS involvement and treatment with interferon-α are independent prognostic factors in Erdheim-Chester disease: a multicenter survival analysis of 53 patients. Blood. 2011; 117: 2778-2782.
- Myra C, Sloper L, Tighe PJ, McIntosh RS, Stevens SE, Gregson RHS, et al. Treatment of Erdheim-Chester disease with cladribine: a rational approach. Br J Ophthalmol. 2004; 88: 844-847.
- Hyman DM, Puzanov I, Subbiah V, Jason E. Faris, Ian Chau, Jean-Yves Blay, et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N Engl J Med. 2015; 373: 726-736.
- Franconieri F, Martin-Silva N, de Boysson H, Galateau-Salle F, Emile JF, Bienvenu B, et al. Superior efficacy and tolerance of reduced doses of vemurafenib plus anakinra in Erdheim-Chester disease: Towards the paradigm of combined targeting and immune therapies. Acta Oncol. 2016; 55: 930-932.
- Schirmer JH, Thorns C, Moosig F, Holle JU. Treatment failure by canakinumab in a patient with progressive multisystemic Erdheim- Chester disease refractory to anakinra: successful use of vemurafenib. Rheumatology (Oxford). 2015; 54: 1932-1934.
- Grumbine FL, Aderman C, Vagefi MR, Kersten RC. Orbital MRI Pre- and Post-Vemurafenib Therapy for Erdheim-Chester Disease. Ophthal Plast Reconstr Surg. 2015; 31: e169.
- Haroche J, Cohen-Aubart F, Emile JF, Maksud P, Drier A, Tolédano D, et al. Reproducible and sustained efficacy of targeted therapy with vemurafenib in patients with BRAF(V600E)-mutated Erdheim-Chester disease. J Clin Oncol. 2015; 33: 411-418.
- Mazor RD, Manevich-Mazor M, Kesler A, Aizenstein O, Eshed I, Jaffe R, et al. Clinical considerations and key issues in the management of patients with Erdheim-Chester Disease: a seven case series. BMC Med. 2014; 12: 221.
- Cohen-Aubart F, Emile JF, Maksud P, Galanaud D, Idbaih A, Chauvet D, et al. Marked efficacy of vemurafenib in suprasellar Erdheim-Chester disease. Neurology. 2014; 83: 1294-1296.