Case Report
Aortic Emergency in Loeys-Dietz Syndrome: Clinical Findings and Molecular Characterization in a Child
Li Y*, Chang Q, Qian X, Sun X and Luo M
Department of Cardiovascular Surgery, National Center for Cardiovascular Diseases and Fuwai Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, China
*Corresponding author: Yan Li, Department of Cardiovascular Surgery, National Center for Cardiovascular Diseases and Fuwai ospital, Chinese Academy of Medical Sciences and Peking Union Medical College, 167 Beilishi Rd, Beijing, 100037 China
Published: 16 Jul, 2016
Cite this article as: Li Y, Chang Q, Qian X, Sun X, Luo M. Aortic Emergency in Loeys-Dietz Syndrome: Clinical Findings and Molecular Characterization in a Child. Clin Surg. 2016; 1: 1060.
Abstract
Loeys-Dietz syndrome, a rare autosomal dominant disorder showing the involvement of cutaneous, cardiovascular, craniofacial, and skeletal systems, presents early in life with rapidly progressive aortic aneurysmal disease. Aortic emergency in young children is an extremely rare occurrence. We present clinical findings and molecular characterization of a 5-year-old Loeys-Dietz syndrome patient whose diagnosis was missed and consequently underwent urgent aortic repair due to aortic rupture. Pediatricians and surgeons should be educated about this rare disorder, to ensure early proper diagnoses resulting in a timely and efficient management strategy before emergence of aortic rupture.
Abbreviations
LDS: Loeys-Dietz Syndrome; TGF: Transforming Growth Factor; TGFBR2: TGF Beta-Receptor Type II; VEDS: Vascular Ehlers-Danlos Syndrome; MFS: Marfan Syndrome
Introduction
Loeys-Dietz syndrome (LDS) is a rare autosomal dominant disorder showing the involvement of cutaneous, cardiovascular, craniofacial, and skeletal systems [1]. In particular, LDS patients show arterial tortuosity with widespread vascular aneurysm and dissection, and have a high risk of aortic dissection or rupture at an early age and at aortic diameters that ordinarily are not predictive of these events [1,2]. LDS is due to mutations in the Transforming Growth Factor (TGF) beta-receptor type I (TGFBR1) and type II (TGFBR2) genes, and has been subdivided in LDS type I (LDSI) and type II (LDSII) on the basis of the presence or the absence of cranio-facial involvement, respectively [1,2]. Furthermore, LDSII patients display at least two of the major signs of vascular Ehlers-Danlos syndrome (vEDS) [2].
Case Presentation
Clinical findings
A 5-year-old boy was referred to emergency room for back pain between the scapulae, hoarseness,
and dyspnea. He was born at term after uncomplicated pregnancy from non-consanguineous parents.
At the age of 4 years he was diagnosed as thoracic aortic aneurysm and Marfan syndrome (MFS).
Clinical follow-up was recommended with consideration of the young age and the unsufficient
aortic size. At the age of 5 years he showed: height 122 cm, arm span 118 cm, upper: lower segment
ratio 0.85. A physical examination revealed bifid uvula, muscular hypotrophy, hypoplastic alae nasi,
microretrognathia, high-arched palate, joint hypermobility (Beighton score 8/9), and velvety texture
skin. Echocardiography demonstrated a large pseudo-aneurysm (73 x 54 mm) at the aortic isthmus
and severe depression of the pulmonary trunk. Therefore, a CT imaging was made rapidly, which
clarified disruption of aortic isthmus vessel wall, formation of a large pseudo aneurysm (diameter 77
mm), and depression of the pulmonary trunk and left pulmonary artery (Figure 1). Those findings
led to the diagnosis of LDSII. An urgent aortic operation was performed. The patient recovered
rapidly and was discharged on the ninth postoperative day. Histopathological examination of the
aortic wall showed diffuse degeneration and elastin fragmentation in the media.
Molecular characterization
Sequencing analysis of genomic DNA disclosed in exon 4 of the TGFBR2 gene the c.1067G>C
missense mutation, leading to the substitution of a glycine with a cytosine in position 356 of the
amino acidic sequence (p.Arg356Pro variation). This novel missense mutation was not detected in
the patient's parents, suggesting that it arose as a de novo event. The Arg356Pro variation, located
in the highly conserved serine-threonine kinase domain of TGFBR2,
was identified only in the patient with LDS but not in 20 MFS patients.
In addition, the Arg356Pro mutation was absent in the 50 healthy
controls. In silico prediction algorithms, polymorphism phenotyping
v2 (PolyPhen-2), characterized the p.Arg356Pro substitution as
damaging and affecting protein function. Furthermore, the TGFBR2
gene c. 1067 G>C missense mutation was validated by Sanger
sequencing (Figure 2).
Figure 1
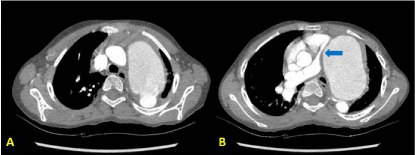
Figure 1
(A) Disruption of aortic isthmus vessel wall; (B) Huge pseudo
aneurysm of the thoracic descending aorta and depression of the pulmonary
trunk and left pulmonary artery (arrow).
Discussion
LDS was first described in 2005 as autosomal dominant with
widespread systemic involvement. The disease is characterized by the
triad of the following: 1) arterial tortuosity, aneurysms, or dissections,
2) hypertelorism, and 3) bifid uvula or cleft palate [1,2]. The patient
showed suggestive signs (i.e., marfanoid habitus without fulfilled
Ghent criteria for MFS, facial, skeletal, cutaneous and cardiovascular
involvement) consistent with LDSII phenotype. In particular, the
patient resembled vEDS for skin presentation and joint involvement,
that generally is less evident in vEDS than in other EDS types and in
LDS patients [2]. However, the straight involvement of the thoracic
aorta of Patient 2 is more suggestive of an LDS rather than a vEDS
phenotype, since in vEDS the medium-sized arteries are more
frequently affected than aorta and great vessels [3].
In the TGFBRs-related disorders no genotype-phenotype
correlation has been drawn, and the prediction of the clinical effects
of different TGFBRs mutations remains difficult to envisage [1,4].
All the clinical entities caused by mutations in TGFBRs genes should
be considered a phenotypic continuum of the same disease, ranging
from isolated thoracic aneurysm to syndromic vascular rupture, and
LDS should be considered a complex syndrome characterized by
full penetrance, pleiotropic effects, and high incidence of de novo
events. On the other hand, LDS shows marked overlapping signs with
other genetically triggered disorders, due to different gene defects
and characterized by vascular fragility, such as MFS, vEDS, and
arterial tortuosity syndrome [2,5]. The differential diagnosis between
these genetically triggered aortic diseases is important because
the progression and prognosis of aortic and branch vessel disease
is significantly different. Although the TGFBRs-related disorders
share genetic mutations in the same signaling pathway, previous
studies have reported that vascular disease in LDS seems to be more
widespread and aggressive than it is in MFS. For these reasons, earlier
surgical intervention should be considered for young LDS patients
with smaller aortic diameter as opposed to the general population
and even Marfan patients. One of our most important findings is the
evidence for the aggressive nature of the aortic pathology in LDS.
Fatal aortic catastrophes occurred in the 5-year-old patient, a clinical
scenario that is extremely rare in Marfan syndrome. This finding
indicates that not only should surgical intervention be considered
earlier, but these patients require strict, aggressive, lifelong
surveillance of the entire arterial tree to identify problems that are
generally amenable to surgical intervention.
Figure 2
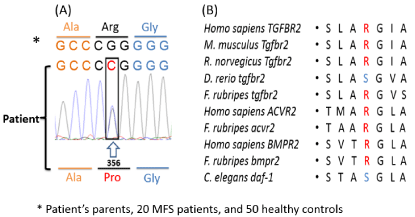
Figure 2
Molecular characterization of the patient by TGFBR2 sequencing
on genomic DNA, obtained from whole peripheral blood. (A) Sequence
chromatogram of the patient, showing the position of the heterozygous
c.1067G>C transversion in TGFBR2 exon 4, leading to the p.Arg356Pro
missense mutation; (B) This mutation occurred at an evolutionally conserved
(red) or chemically similar (blue) amino acid in serinethreonine kinase domain
in mouse, rat, zebrafish (Tgfbr2), human, pufferfish (Acvr2), and nematode
(daf proteins).
Conclusion
We present a 5-year-old patient with LDS who was only correctly diagnosed after aortic emergency, and report an Arg356Pro mutation in the TGFBR2 gene. Pediatricians and surgeons should be aware of this rare disorder, to ensure early proper diagnoses resulting in a timely and efficient management strategy before emergence of aortic rupture.
References
- Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005, 37: 275-281.
- Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006, 355: 788-798.
- Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med. 2000, 342: 673-680.
- Stheneur C, Collod-Béroud G, Faivre L, Gouya L, Sultan G, Le Parc, et al. Identification of 23 TGFBR2 and 6 TGFBR1 gene mutations and genotype-phenotype investigations in 457 patients with Marfan syndrome type I and II, Loeys-Dietz syndrome and related disorders. Hum Mutat. 2008, 29: 284-295.
- Sakai H, Visser R, Ikegawa S, Ito E, Numabe H, Watanabe Y, et al. Comprehensive genetic analysis of relevant four genes in 49 patients with Marfan syndrome or Marfan-related phenotypes. Am J Med Genet A. 2006, 140: 1719-1725.