Research Article
Adrenocortical Carcinoma in Adults and Children: A Population-Based Outcomes Study involving 1,623 Patients from the Surveillance, Epidemiology, and End Result (SEER) Database (1973-2012)
Mahendraraj K1, Lau CSM1,2, Sidhu K1 and Chamberlain RS1-3*
1Department of Surgery, Saint Barnabas Medical Center, NJ, USA
2Saint George's University School of Medicine, West Indies
3Department of Surgery, New Jersey Medical School, Rutgers University, NJ, USA
*Corresponding author: Ronald S. Chamberlain, Chairman and Surgeon-in-Chief, Department of Surgery, Saint Barnabas Medical Center, Professor of Surgery, Rutgers University, New Jersey Medical School (NJMS) 94 Old Short Hills Rd., Livingston, New Jersey 07039
Published: 06 May, 2016
Cite this article as: Mahendraraj K, Lau CSM, Sidhu
K, Chamberlain RS. Adrenocortical
Carcinoma in Adults and Children: A
Population-Based Outcomes Study
involving 1,623 Patients from the
Surveillance, Epidemiology, and End
Result (SEER) Database (1973-2012).
Clin Surg. 2016; 1: 1017.
Abstract
Background: Adrenocortical carcinoma (ACC), a rare endocrine tumor, is typically aggressive in adult patients, while ACC inpediatric patients follows an unpredictable course which is poorly
studied. This study examines a large cohort of adult and pediatric ACC patients in an effort to
identify demographic, pathologic, and clinical factors which affect clinical outcomes.
Methods: Data on 1,623 patients (101 pediatric patients <20 years and 1,522 adult patients ≥20
years) were abstracted from the Surveillance, Epidemiology, and End Result (SEER) database (1973-
2012). Standard statistical analyses were performed.
Results: ACC most commonly occurred in Caucasian females with tumors>4 cm in size. More
pediatric patients age <5 were Hispanic compared to adults. Pediatric patients age <5 had
significantly less metastatic disease compared to patients age 5-9, age 10-19 and age ≥20. Surgical
resection was the most common treatment modality and significantly improved overall survival
(OS) in all patients. Pediatric patients age<5 had the highest OS and lowest mortality whereas,
pediatric patients age 10-19 had the highest mortality and the lowest 1-, 2- and 5-year survival.
Multivariate analysis identified age>10 (OR 46.6), distant disease (OR 13.7) and undifferentiated
grade (OR 6.0) as independently associated with increased mortality, p <0.05.
Conclusions: ACC is an aggressive tumor affecting adults and children with a bimodal distribution.
Surgical resection significantly improves OS in all groups, particularly pediatric patients age <5.
Advancing age represents a key factor in the prognosis of ACC and should be considered in addition
to tumor grade and stage, when risk stratifying patients.
Keywords: Adrenocortical carcinoma; Adrenal cortex tumors; Pediatric cancer; SEER
Introduction
Adrenocortical carcinoma (ACC) is a rare and aggressive tumor which peaks in incidence
during the first and fifth decades of life. ACC has an estimated worldwide annual incidence of 2
cases per million overall. The highest reported annual incidence of pediatric ACC in the world
occurs in Southern Brazil (3.4 per million) and is 10-15 fold higher compared to the United States
(U.S.), where the annual incidence of pediatric ACC is 0.2-0.3 cases per million, with approximately
25 new cases diagnosed annually [1,2]. This marked difference in ACC incidence between these
two countries has been attributed to a higher prevalence of p53 mutations among the Brazilian
population, including TP53 polymorphisms and overexpression of polo-like kinase 1 (PLK1) which
downregulates p53 activity [1-6].
Pediatric ACC is a distinct disease separate from adult onset ACC, that follows a highly
unpredictable course [7,8]. Unlike the adult population, the majority of pediatric ACCs are
functional tumors and present with symptoms secondary to hypersecretion of cortisol, aldosterone,
or androgens. These tumors often manifest as Cushing syndrome and/or virilization. Up to 50%
of pediatric ACCs are attributed to defined genetic alterations and associated with congenital
syndromes such as Li-Fraumeni and Beckwith-Weidmann syndrome
[4]. Weiss et al. [9] have described a pathological criteria for
predicting poor prognosis among adult ACC patients: nuclear grades
III or IV, mitotic rate >5/50 high-power field (hpf), atypical mitoses,
clear cells comprising ≤25% of the tumor, diffuse architecture,
microscopic necrosis, and vascular or capsular invasion. Tumors
having three or more of these criteria exhibited aggressive clinical
behavior [1,9]. No definitive pathologic criterion has been described
in the pediatric population given the rarity of this disease [10]. The
majority of the existing literature on pediatric ACCs is derived from
the Brazilian population, while most American studies are limited to
single institution reviews with small sample size [10-12].
ACC in the pediatric population is rare and current knowledge
regarding pediatric ACC is limited since very few studies have
examined treatment approaches and outcomes in children. This
study sought to examine a large cohort of adult and pediatric ACC
from the Surveillance, Epidemiology, and End Results (SEER)
database in an effort to identify demographic, clinical, and treatment
strategies which impact clinical outcomes and potentially guide
therapeutic decision making and assist in clinical trial development
and appropriate accrual.
Methods
Demographic and clinical data for the current study was extracted from the Surveillance, Epidemiology, and End Result (SEER) database provided by the National Cancer Institute between 1973 and 2012 (Figure 1). SEER Stat software version 8.0.4 was utilized to extract data from 18 SEER registries (Alaska Native Tumor Registry, Arizona Indians Cherokee Nation, Connecticut, Detroit, Georgia Center for Cancer Statistics, Greater Bay Area Cancer Registry, Greater California, Hawaii, Iowa, Kentucky, Los Angeles, Louisiana, New Jersey, New Mexico, Seattle-Puget Sound, and Utah). 1,623 patients with histologically confirmed ACC were identified and exported to IBM SPSS v20.2. Patients with a primary diagnosis of ACC were identified to form the final study cohort, using the SEER International Classification of Disease for Oncology (ICD-0-3) code 194.0. Demographic and clinical data extracted included: age, gender, race, tumor size, stage, grade, lymph node involvement, and type of treatment received (surgery, radiation, both, or no treatment). SEER summary staging was used to define the extent of disease for all cases, and was outlined as localized (confined to adrenal gland), regional (invasion of adjacent structures or lymph node involvement), or distant (metastatic) disease. Two subgroups were created based on age; Pediatric patients were defined as age <20 years and adult patients defined as age ≥20 years. Pediatric patients were further divided into three age groups; '< 5', '5-9' and '10-19' years. Endpoints examined included overall mortality, cancer specific mortality, means overall survival, survival by treatment, and cancer-specific 1-, 2- and 5- year survival. The Kaplan–Meier method was used to develop the survival curves and estimate mean overall and cancer specific survival. Categorical variables were compared between the two groups using the Chi square test, and continuous variables were compared using analysis of variance (ANOVA). Multivariate analysis using the backward Wald method was performed to calculate odds ratios (OR) and to determine independent factors affecting mortality. Missing or unknown data were excluded from multivariate analysis. Kaplan- Meier analysis was used to compare long term actuarial survival between the groups. Statistical significance was defined as p <0.05.
Figure 1
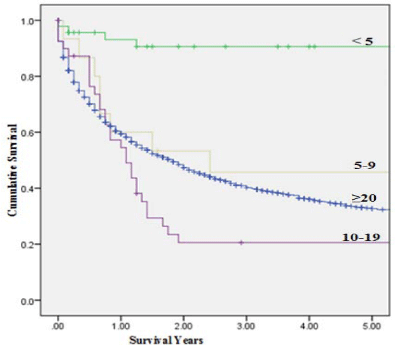
Figure 1
Kaplan-Meier Estimate of 5-Year Survival for 1,602 Adult
patients and 101 Pediatric Patients with Adrenocortical Carcinoma from the
Surveillance Epidemiology and End Result (SEER) Database (1973-2012).
Table 1
Table 1
Demographic Profile of 1,522 Adult and 101 Pediatric Patients with Adrenocortical Carcinoma from the Surveillance Epidemiology and End Result (SEER) Database (1973-2012).
Table 2
Table 2
Demographic Profile of 1,522 Adult and 101 Pediatric Patients with Adrenocortical Carcinoma from the Surveillance Epidemiology and End Result (SEER) Database (1973-2012).
Results
A total of 1,623 ACC cases were identified between 1973 and
2012, of which 101 (6.2%) were pediatric and 1,522 (93.8%) were
adults. Among pediatric subgroups; 45.5% (N=46) were age <5 years,
14.9% (N=15) were between age 5-9 years, and 39.6% (N=40) were
between ages 10 to 19 years (Table 1).
Demographic characteristics
The mean age was 7.8±6.7 years among pediatric patients and
55±15.2 years in adult patients (Table 1). The majority of ACC
occurred in females across all age groups; <5 years (67.4%), 5-9 years
(60.0%), 10-19 years (67.5%) and >20 years (55.6%).The majority of
ACC cases occurred in Caucasians (77.4%), followed by Hispanics
(9.4%, N=152), African Americans (6.8%; N=111), and Asian, Pacific
Islander or Native Americans (6.0%, N=98). Ethnicity was not
reported in 5 patients (0.3%). Significantly more pediatric patients<
5 years (30.4%) were Hispanics compared to those 5-9 year of age
(13.3%), 10-19 years (17.5%), and adults (8.5%), p<0.001.
Tumor characteristics
Across all age groups, most patients presented with tumor size > 4
cm (overall; 72.0%) (Table 2). The majority of the overall ACCgroup
presented with localized disease (40.8%; N=415), followed by regional
(18.9%; N=192) and distant disease (33.5%; N=341). Sixty nine
cases (6.8%) of ACC were unstaged. Pediatric patients<5 years had
significantly lower distant disease rates (7.1%; N=2), compared to
those age 5-9 years (42.9%; N=6), 10-19 years (57.7%; N=15), and >20
years (33.5%; N=318), p=0.003. The majority of children<5 years had
localized disease (75.0%; N= 21) compared to all other age groups,
p=0.002.
Treatment
Overall, 67.1% of ACC patients were managed with surgical
resection alone (N=1,089), 3.3% of patients received radiotherapy
alone (N=54), and 6.8% received combination surgery and radiation
(N=111). 324 (20.0%) patients had no treatment while the treatment
modality of 2.8% was unknown (N=45) (Table 3). Surgical resection
was most commonly performed in the pediatric patients<5 (93.5%;
N=43) compared to those 5-9 years (80.0%, N=12), 10-19 years
(57.5%; N=23) and adults (66.4%; N=1,011; p=0.068). Pediatric
patients age 10-19 years received multimodality surgery and radiation
therapy at the highest rate (12.5%; N=5) compared to adults (6.9%;
N=105) and pediatric patients 5-9 years of age (6.7%; N=1).
Clinical outcomes and survival
Overall and cancer specific mortality among the entire ACC cohort
was 69.3% and 62.2%, respectively. Mean overall survival (OS) for
patients<5 (25.7±2.0 years) was significantly better than for patients
age 5-9 (13.3±4.4 years), age 10-19 (8.3±2.5 years) and adults (7.6±0.4
years); p <0.001. Pediatric patients<5 years had lower overall (13.0%,
N=6) and cancer-specific mortality (9.1%; N=4) as well as higher 1-,2-
and 5-year survival (93%, 90% and 90% respectively) compared to
all other groups; p <0.001. Alternatively, pediatric patients age 10-19
years had the highest overall and cancer-specific mortality (72.5% and
72.5%) and the lowest 1-, 2- and 5-year survival (58%, 21% and 21%
respectively) among all age groups, p<0.001. Surgical management
improved survival in all age groups; pediatric patients<9 years
(30.6±2.1), 10-19 years (5.7±1.8) and adults (13.6±0.8), p<0.001.
Patients aged 10-19 years had the longest survival with multimodality
surgery and radiation therapy compared to surgery alone (15.5±7.9
years versus 5.7±1.8 years respectively, p <0.001).
Multivariate analysis
Multivariate analysis identified age >10 (OR 46.6, CI=2.0-
1, 111.0), distant disease (OR 13.7, CI=1.6-121.5) and undifferentiated
grade (OR 6.0, CI=1.2-29.5) as independently associated with
increased mortality, p <0.05. These same factors were significant in
both the pediatric and adult ACC groups. Hispanic race (OR 0.66,
CI=0.46-0.93) was associated with a decreased mortality on univariate analysis, p <0.001.
Discussion
Pediatric ACC is a rare tumor comprising only 0.2% of all
childhood malignancies, with the highest incidence rates of 0.4 per
million occurring during the first 4 years of life [4]. The incidence
of ACC varies from 0.2 per million in the U.S. to as high as 3.4 per
million in Southern Brazil where there is an increased prevalence
of TP53 polymorphisms and overexpression of polo-like kinase 1
(PLK1) which downregulates p53 activity [1,2,5,6,13,14].
ACC occurs most commonly among Caucasian females, with
a greater female preponderance among younger children, which is
consistent with previous studies [12,14,15]. Michalkiewicz et al. [15]
conducted a retrospective study involving 254 pediatric patients
(<20 years) from the International Pediatric Adrenocortical Tumor
Registry, and reported a female predominance in age groups 0-3 years
(1.7:1) and ≥13 years (6.2:1), however no such significant gender
difference was noted for the 4-12 year old age group (0.8:1). Similarly,
McAteer et al. [12] conducted a SEER database study involving
85 pediatric ACC patients <20 years of age and reported a female
predominance in all age groups except for children 10-14 years of age
(1:1). The authors also reported that 91.8% of pediatric ACC occurred
among Caucasians and 5.9% in Asians [12]. Pediatric patients <5
years of age presented far more often with localized disease (75%),
whereas pediatric patients >5 years of age and adults often presented
with advanced disease (42.9%, 57.7%, and 33.5% of patients 5-9, 10-
19, and >20 years, respectively). Similarly, McAteer et al. [12] have
previously reported a retrospective study involving 85 pediatric
patients <20 years of age and similarly reported that children ≥5
years presented with higher rates of distant disease (43.0-67.0%)
compared to children <5 years (11.0%), p=0.02 [12]. Additionally,
these authors reported that the majority of patients presented with
tumors >10 cm in size across all age groups except for children <5
years [12]. Advanced disease presentation among older children and
adults were attributed primarily to differences in tumor presentation
among different age groups. Several studies have reported that >80%
of pediatric ACC are associated with virilization, due to marked
overproduction of androgens [4,16]. Hanna et al. [17] conducted
a retrospective review of 23 patients <19 years of age and reported
that 61% of patients presented with hormonal signs and symptoms,
26% with an abdominal mass, and 13% with abdominal pain. The
functional nature of pediatric ACC permits earlier detection, usually
within 5-8 months of symptom occurrence, and improved survival
[12]. In contrast, >50% of adult ACC patients presented with no
recognizable endocrine syndromes but instead reported vague
symptoms such as abdominal pain or fullness. The lack of symptoms
in adolescents and adults often leads to a significant delay in diagnosis
permitting tumor growth and increased likelihood of metastases,
likely resulting in worse outcomes and increased mortality [16,18].
Radical adrenalectomy is the primary therapy for ACC and
complete resection has been shown to improve long term survival and
prevent recurrence [19,20]. Numerous studies have demonstrated
5-year survival rates ranging from 32% to 48% for patients who
undergo a complete resection [13,21]. In contrast, patients who
undergo incomplete resection of the primary neoplasm have a poor
prognosis with median survival times of <1 year [22]. Margonis et al.
[23] reviewed 165 patients from a multi-institutional database and
reported a significantly prolonged survival in patients achieving R0
surgical margins (median survival of 96.3 months vs. 25.1 months,
p<0.001; 5-year OS of 64.8% vs. 33.8%, p <0.001) compared to patients
with R1 surgical margins.
Given the overall poor prognosis of ACC even when surgical
resection is performed, the use of adjuvant therapy has been
extensively investigated. While radiotherapy studies have yielded
variable success in adult patients, its role in pediatric ACC is ill
defined and under investigated [24]. In the current study, radiation
alone did not improve survival, however multimodality therapy with
surgery and radiation improved survival among pediatric patients
aged 10-19 years compared to surgery alone, possibly attributable
to higher rates of advanced disease and lymph node involvement
reported among older children. Saboltch et al. [25] conducted a
retrospective study involving 40 ACC patients with localized disease
(20 patients receiving adjuvant radiation therapy after surgery and 20
patients receiving surgical resection alone) and reported significantly
improved local control with postoperative radiation compared to
surgical resection alone (recurrence rates of 5% vs. 60%, p=0.0005).
Although the patients were matched for stage and grade, tumor size
(mean of 12.6cm vs. 10.6cm, p=0.72) and negative margins (70% vs.
55%, p >0.05) were variable slightly between the adjuvant radiation
and surgical resection alone groups [25].
Mitotane (dichlorodiphenildichloroethane) alone or in
combination has emerged as the chemotherapy drug of choice in
the setting of metastatic or localized advanced or aggressive ACC
disease. The FIRM-ACT group (2012) randomized 304 patients with
advanced ACC not amenable to radical surgical resection to receive
either etoposide, doxorubicin, cisplantin (EDP) and mitotane or
streptozocin and mitotane [26]. Patients receiving EDP-mitotane
experienced significantly higher response rates (23.2% vs. 9.2%,
p<0.001) and longer median progression-free survival (PFS) (5.0
months vs. 2.1 months, HR 0.55, p<0.001), compared to patients
receiving streptozocin and mitotane [26]. No significant difference
in OS was observed (14.8 months vs. 12.0 months, HR 0.79, p=0.07)
[26]. Postlewait et al. [27] studied 207 ACC patients with high risk
features including tumor rupture, positive margins, lymph node
involvement, high grade and advanced stage and reported that
adjuvant mitotane following surgical resection was not associated
with improved recurrent-free survival (RFS) or OS after accounting
for stage and adverse tumor and treatment-related factors [27].
The authors noted that patients with an R1 resection were more
likely to receive adjuvant mitotane (OR=1.4, p=0.30) compared
to those achieving R0 resection [27]. Patients with stage IV disease
was associated with increased use of adjuvant mitotane (OR=6.3,
p=0.03) on univariate analysis, but not multivariate analysis (OR=4.8, p=0.07) [27]. Furthermore, R1 resection (HR=2.8, p=0.002) and
R2 resection (HR=4.4, p=0.002) was independently associated with
reduced recurrent-free and overall survival [27]. A trend towards
stage IV disease associated with reduced recurrent-free and overall
survival was also observed (HR=7.9, p=0.05) [27].
Given the difficulty in treating advanced and metastatic ACC,
various molecular target therapies have been investigated. Quinkler
et al. [28] conducted a retrospective study involving 10 patients with
advanced ACC treated with the epidermal growth factor receptor
(EGFR) inhibitor, erlotinib, and failed to demonstrate any clinical
efficacy. Wortmann et al. [29] conducted a small cohort evaluated
the use of bevacizumab, a monoclonal antibody against vascular
endothelial growth factor (VEGF), which is highly expressed in
ACC, and reported progressive disease in all 10 patients [29].
Similarly, sorafenib was employed in combination with paclitaxel
and failed to demonstrate any activity in a cohort of 25 ACC patients
[30]. In contrast, the multi-tyrosine kinase inhibitor, sunitinib, has
demonstrated anti-proliferative effects in vitro and appears to down
regulate HSD3B2, resulting in reduced adrenal steroidogenesis
[31]. Drugs targeting insulin-like growth factor (IGF)-2 have also
been investigated with mixed results [32]. A phase 2 trial evaluating
cixutumumab, an IGF-R1R antibody, has demonstrated limited
therapeutic efficacy in a cohort of 19 ACC patients [33]. Naing et al.
[34] conducted a cohort study involving 26 ACC patients receiving
cixutumumab and temsirolimus and reported that 42% of patients
achieved stable disease for ≥6 months [34].
As with most malignancies, distant disease is associated with poor
prognosis and increased mortality in ACC patients. In addition, age
>10 years was determined to be associated with increased mortality.
McAteer et al. [12] have similarly reported age at diagnosis was the
only predictive factor of poor outcome in their study of 85 pediatric
ACC patients. Ribeiro et al. [35] demonstrated that age >3.5 years
was associated with worse outcomes. Similarly, Michalkiewicz
et al. [15] reported age <3 years was a favorable prognostic factor.
Proposed theories to explain improved outcomes in younger
children affected with ACC include the fact that ACC in young
children arise from the fetal zone of the adrenal cortex, which
represents 85% of the adrenal cortex during fetal development, and
produces dehydroepiandrosterone resulting in virilization [8,15].
Although increased tumor size has been reported to be associated
with decreased survival in several prior studies, in the current study
nearly 90% of all ACC patients presented with tumors > 4 cm without
demonstrating decreased survival and it was not an independent risk
factor affecting mortality [35-37]. Wieneke et al. [10] studied 83 ACC
pediatric patients (<20 years) and reported that tumors >10.5 cm had
worse outcomes compared to smaller tumors; however, size alone
was similarly not an independent prognostic factor [10]. In addition,
these same authors reported that tumor weight >400gm was a better
predictor of prognosis; however tumor weight did not prove to be an
independent prognostic factor alone [10]. Michalkiewicz et al. [15]
reported tumor weight <200gm, virilization and stage I disease as
favorable prognostic factors [15]. Similarly, Ribeiro et al. [35] analyzed
40 cases of pediatric ACC in Brazil and reported tumor volume >200
cm3 or weight >80gm as associated with worse outcomes.
Among many prognostic factors identified in ACC, the Ki-67
proliferation index or mitotic count, have been shown to be the most
important prognostic factor in both localized and metastatic ACC
[32,38]. ACC generally exhibit a Ki-67 >5% [32]. The German ACC
registry involving 319 ACC patients, demonstrated that Ki-67 was
the most important prognostic factor for recurrence-free survival
(RFS) (HR=1.042, p <0.0001) and OS (HR = 1.051, p <0.0001) [38].
According to the authors, clinical outcome differed significantly
between patients with Ki67<10%, 10-19%, and ≥20% (median RFS,
53.2 vs. 31.6 vs. 9.4 months; median OS, 180.5 vs. 113.5 vs. 42.0
months) [38]. Furthermore, >20 mitotic counts/50 hpf have been
associated with worse prognosis compared to low grade ACC with
<20 mitotic counts/50 hpf [32].
There are several limitations of the current study which should
be taken into account. Firstly, the SEER database does not code for
clinical factors, such as virilization and specific endocrine syndromes
such as Cushing's syndrome. Several tumor factors such as tumor
grade, weight and mitotic index were also unavailable, which may
influence survival. Secondly, information on diagnostic imaging,
chemotherapy, and patient follow up was lacking. Data on surgical
and radiation therapy utilized was available in the SEER database,
however, information on surgical resection margins or chemotherapy
was not, and this limits the study's ability to comment on the impact
of adjuvant or neoadjuvant therapy. There also may be an element of
selection bias, since SEER registries are more likely to sample from
urban than from rural areas. Despite these limitations, the SEER
database has data obtained from 26% of the United States population,
and these findings can be generalized to the overall pediatric and
adult population.
Summary
ACC is a rare aggressive tumor in the pediatric population with an incidence of 0.2 – 0.3 per 1 million persons. Similar to the adult population, ACC occurs most commonly among Caucasian females, however more Hispanic children<5 years were affected. Children <5years of age often present with hormonal signs and symptoms such as virilization, permitting earlier detection and treatment. Children >5 years of age as well as adults typically present with nonspecific signs and symptoms, resulting in delayed diagnosis, larger tumors and more advanced disease. Surgery is the preferred treatment modality and significantly prolongs survival in patients achieving an R0 resection. Adjuvant mitotane alone or in combination is the current gold standard for patients with advanced disease but results are mixed. Molecular target therapy has been increasingly studied, however, limited success has been demonstrated to date. Despite many published prognostic factors for ACC, Ki-67 immunomarker and mitotic count are the most important predictor of survival for both localized and metastatic ACC. Given the limited number of patients who received adjuvant radiation therapy no conclusions can be drawn about its use. Additional research into the role of adjuvant therapy in both pediatric and adult ACC patients is needed to develop more efficacious and targeted therapy regimens for patients with ACC.
References
- Rodriguez-Galindo C, Figueiredo BC, Zambetti GP, Ribeiro RC. Biology, clinical characteristics, and management of adrenocortical tumors in children. Pediatr Blood Cancer. 2005; 45: 265-273.
- Schteingart DE, Doherty GM, Gauger PG, Giordano TJ, Hammer GD, Korobkin M, et al. Management of patients with adrenal cancer: recommendations of an international consensus conference. Endocr Relat Cancer. 2005; 12: 667-680.
- Ribeiro RC, Sandrini F, Figueiredo B, Zambetti GP, Michalkiewicz E, Lafferty AR, et al. An inherited p53 mutation that contributes in a tissuespecific manner to pediatric adrenal cortical carcinoma. Proc Natl Acad Sci U S A. 2001; 98: 9330-9335.
- Ribeiro RC, Figueiredo B. Childhood adrenocortical tumours. Eur J Cancer. 2004; 40: 1117-1126.
- Heinze B, Herrmann LJ, Fassnacht M, Ronchi CL, Willenberg HS, Quinkler M, et al. Less common genotype variants of TP53 polymorphisms are associated with poor outcome in adult patients with adrenocortical carcinoma. Eur J Endocrinol. 2014; 170: 707-717.
- Bussey KJ, Bapat A, Linnehan C, Wandoloski M, Dastrup E, Rogers E, et al. Targeting polo-like kinase 1, a regulator of p53, in the treatment of adrenocortical carcinoma. Clin Transl Med. 2016; 5: 1.
- Paton BL, Novitsky YW, Zerey M, Harrell AG, Norton HJ, Asbun H, et al. Outcomes of adrenal cortical carcinoma in the United States. Surgery. 2006; 140: 914-920.
- Dehner LP, Hill DA. Adrenal cortical neoplasms in children: why so many carcinomas and yet so many survivors? Pediatr Dev Pathol. 2009; 12: 284-291.
- Aubert S, Wacrenier A, Leroy X, Devos P, Carnaille B, Proye C, et al. Weiss system revisited: a clinicopathologic and immunohistochemical study of 49 adrenocortical tumors. Am J Surg Pathol. 2002; 26: 1612-1619.
- Wieneke JA, Thompson LD, Heffess CS. Adrenal cortical neoplasms in the pediatric population: a clinicopathologic and immunophenotypic analysis of 83 patients. Am J Surg Pathol. 2003; 27: 867-881.
- Mendonca BB, Lucon AM, Menezes CA, Saldanha LB, Latronico AC, Zerbini C, et al. Clinical, hormonal and pathological findings in a comparative study of adrenocortical neoplasms in childhood and adulthood. J Urol. 1995; 154: 2004-2009.
- McAteer JP, Huaco JA, Gow KW. Predictors of survival in pediatric adrenocortical carcinoma: a Surveillance, Epidemiology, and End Results (SEER) program study. J Pediatr Surg. 2013; 48: 1025-1031.
- Icard P, Chapuis Y, Andreassian B, Bernard A, Proye C. Adrenocortical carcinoma in surgically treated patients: a retrospective study on 156 cases by the French Association of Endocrine Surgery. Surgery. 1992; 112: 972-979.
- Kerkhofs TM, Ettaieb MH, Verhoeven RH, Kaspers GJ, Tissing WJ,Loeffen J, et al. Adrenocortical carcinoma in children: first populationbased clinicopathological study with long-term follow-up. Oncol Rep. 2014; 32: 2836-2844.
- Michalkiewicz E, Sandrini R, Figueiredo B, Miranda EC, Caran E, Oliveira-Filho AG, et al. Clinical and outcome characteristics of children with adrenocortical tumors: a report from the International Pediatric Adrenocortical Tumor Registry. J Clin Oncol. 2004; 22: 838-845.
- Wajchenberg BL, Albergaria Pereira MA, Medonca BB, Latronico AC, Campos Carneiro P, Alves VA, et al. Adrenocortical carcinoma: clinical and laboratory observations. Cancer. 2000; 88: 711-736.
- Hanna AM, Pham TH, Askegard-Giesmann JR, Grams JM, Iqbal CW, Stavlo P, et al. Outcome of adrenocortical tumors in children. J Pediatr Surg. 2008; 43: 843-849.
- Barzilay JI, Pazianos AG. Adrenocortical carcinoma. Urol Clin North Am. 1989; 16: 457-468.
- Ciftci AO, Senocak ME, Tanyel FC, Buyukpamukcu N. Adrenocortical tumors in children. J Pediatr Surg. 2001; 36: 549-554.
- Stewart JN, Flageole H, Kavan P. A surgical approach to adrenocortical tumors in children: the mainstay of treatment. J Pediatr Surg. 2004; 39: 759-763.
- Pommier RF, Brennan MF. An eleven-year experience with adrenocortical carcinoma. Surgery. 1992; 112: 963-970.
- Fishman EK, Deutch BM, Hartman DS, Goldman SM, Zerhouni EA, Siegelman SS. Primary adrenocortical carcinoma: CT evaluation with clinical correlation. AJR Am J Roentgenol. 1987; 148: 531-535.
- Margonis GA, Kim Y, Prescott JD, Tran TB, Postlewait LM, Maithel SK, et al. Adrenocortical Carcinoma: Impact of Surgical Margin Status on Long-Term Outcomes. Ann Surg Oncol. 2016; 23: 134-141.
- Polat B, Fassnacht M, Pfreundner L, Guckenberger M, Bratengeier K, Johanssen S, et al. Radiotherapy in adrenocortical carcinoma. Cancer 2009; 115: 2816-2823.
- Sabolch A, Else T, Griffith KA, Ben-Josef E, Williams A, Miller BS, et al. Adjuvant radiation therapy improves local control after surgical resection in patients with localized adrenocortical carcinoma. Int J Radiat Oncol Biol Phys. 2015; 92: 252-259.
- Fassnacht M, Terzolo M, Allolio B, Baudin E, Haak H, Berruti A, et al. Combination chemotherapy in advanced adrenocortical carcinoma. N Engl J Med. 2012; 366: 2189-2197.
- Postlewait LM, Ethun CG, Tran TB, Prescott JD, Pawlik TM, Wang TS, et al. Outcomes of Adjuvant Mitotane after Resection of Adrenocortical Carcinoma: A 13-Institution Study by the US Adrenocortical Carcinoma Group. J Am Coll Surg. 2016; 222: 480-490.
- Quinkler M, Hahner S, Wortmann S, Johanssen S, Adam P, Ritter C, et al. Treatment of advanced adrenocortical carcinoma with erlotinib plus gemcitabine. J Clin Endocrinol Metab. 2008; 93: 2057-2062.
- Wortmann S, Quinkler M, Ritter C, Kroiss M, Johanssen S, Hahner S, et al. Bevacizumab plus capecitabine as a salvage therapy in advanced adrenocortical carcinoma. Eur J Endocrinol. 2010; 162: 349-356.
- Berruti A, Sperone P, Ferrero A, Germano A, Ardito A, Priola AM, et al. Phase II study of weekly paclitaxel and sorafenib as second/third-line therapy in patients with adrenocortical carcinoma. Eur J Endocrinol. 2012; 166: 451-458.
- Kroiss M, Reuss M, Kuhner D, Johanssen S, Beyer M, Zink M, et al. Sunitinib Inhibits Cell Proliferation and Alters Steroidogenesis by Down-Regulation of HSD3B2 in Adrenocortical Carcinoma Cells. Front Endocrinol (Lausanne). 2011; 2: 27.
- Libe R. Adrenocortical carcinoma (ACC): diagnosis, prognosis, and treatment. Front Cell Dev Biol. 2015; 3: 45.
- Lerario AM, Worden FP, Ramm CA, Hesseltine EA, Stadler WM, Else T, et al. The combination of insulin-like growth factor receptor 1 (IGF1R) antibody cixutumumab and mitotane as a first-line therapy for patients with recurrent/metastatic adrenocortical carcinoma: a multi-institutional NCI-sponsored trial. Horm Cancer. 2014; 5: 232-239.
- Naing A, Lorusso P, Fu S, Hong D, Chen HX, Doyle LA, et al. Insulin growth factor receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with metastatic adrenocortical carcinoma. Br J Cancer. 2013; 108: 826-830.
- Ribeiro RC, Sandrini Neto RS, Schell MJ, Lacerda L, Sambaio GA, Cat I. Adrenocortical carcinoma in children: a study of 40 cases. J Clin Oncol. 1990; 8: 67-74.
- Bugg MF, Ribeiro RC, Roberson PK, Lloyd RV, Sandrini R, Silva JB, et al. Correlation of pathologic features with clinical outcome in pediatric adrenocortical neoplasia. A study of a Brazilian population. Brazilian Group for Treatment of Childhood Adrenocortical Tumors. Am J Clin Pathol. 1994; 101: 625-629.
- Sandrini R, Ribeiro RC, DeLacerda L. Childhood adrenocortical tumors. J Clin Endocrinol Metab 1997; 82: 2027-2031.
- Beuschlein F, Weigel J, Saeger W, Kroiss M, Wild V, Daffara F, et al. Major prognostic role of Ki67 in localized adrenocortical carcinoma after complete resection. J Clin Endocrinol Metab. 2015; 100: 841-849.