Research Article
Rare Case of Inflammatory Myofibroblastic Tumor of the Adrenal Mimicking Adrenocortical Carcinoma
Goonj Johri1, Gyan Chand1, Mishra SK1*, Paramita Paul2 and Vinita Agrawal2
1Department of Endocrine Surgery, SGPGIMS, India
2Department of Pathology, SGPGIMS, India
*Corresponding author: Mishra SK, Department of Endocrine Surgery, Sanjay Gandhi PG Institute of Medical Sciences, Lucknow, India
Published: 28 Jun, 2017
Cite this article as: Johri G, Chand G, Mishra SK, Paul P,
Agrawal V. Rare Case of Inflammatory
Myofibroblastic Tumor of the Adrenal
Mimicking Adrenocortical Carcinoma.
Clin Surg. 2017; 2: 1524.
Abstract
Background: Inflammatory Myofibroblastic tumors (IMT) arising from the adrenal gland are
extremely rare. Until now only 5 cases have been reported in literature.
Case Report: 55 year old male under evaluation for fever, right upper abdominal pain and nonprogressive
jaundice on abdominal CECT was found to have a large right suprarenal mass, high
suspicion of adrenocortical carcinoma. He underwent adrenalectomy after hormonal evaluation.
Final histopathology was Inflammatory Myofibroblastic tumor. A rare tumor composed of plump,
spindle cells or myofibroblasts and histiocytoid cells arranged haphazardly and in fascicles,
accompanied by prominent chronic inflammatory infiltrate, particularly plasma cells.
Conclusion: IMT of the adrenal are rare. Radiologically and clinically they mimic a malignant
process and must be considered in the differential of radiologically suspicious adrenal masses.
Complete surgical resection is mandatory as malignancy cannot be excluded preoperatively.
Keywords: Inflammatory myofibroblastic tumors; Adrenal gland; Histopathology;
Immunohistochemistry
Background
Inflammatory Myofibroblastic tumors (IMT) are an uncommon spindle-cell proliferation that may involve any site - abdomen, lungs, orbit, gastrointestinal and genitourinary tracts [1]. Usually seen in children and young adults. The advent of immunohistochemistry brought increasing awareness of its distinctive features [2]. The adrenal gland is a rare primary site, with only four reports in the world literature [1,3-5]. Clinical differential diagnoses include adrenocortical tumours, phaeochromocytoma, adrenal metastases and tuberculosis. Imaging features are generally nonspecific, preoperative differentiation from other adrenal masses is very difficult. Complete surgical resection is therefore mandatory as malignancy cannot be excluded preoperatively. We report a fifth case of this rare entity.
Case Presentation
55 year old, male, from a small town in India was being evaluated for fever, right upper abdominal
pain and jaundice of 14 days duration. He underwent an Ultrasonography of abdomen which
showed large liver SOL and was referred to the Gastroenterology clinic at our Institute. Fever was
high grade, intermittent type, not associated with chills and rigor. Upper abdominal pain was acute
onset, right sided, continuous dull aching in nature. No radiation, shifting or referred pain. Jaundice
was non progressive, associated with loss of appetite, nausea and malaise. Not associated with clay
colored stools, pruritis or abdominal distension. No history of loss of weight, hematemesis or melena.
No history of similar illness in the past. No history of Tuberculosis or any other chronic illness.
He was recently diagnosed with Hypertension. No history of smoking or alcohol. General physical
examination was unremarkable except for icterus. Abdominal examination revealed hepatomegaly.
Lower border of liver was palpable 5 cm below the right costal margin. No other organomegaly or
mass abdomen palpable. Blood work up showed raised total and direct bilirubin with raised liver
enzymes and normal coagulation parameters and viral serology. Patient was managed on the lines
of acute viral hepatitis and liver functions returned to normal levels over two weeks with supportive
treatment. To characterise the, a CECT Abdomen (Figure 1) was done which showed 17.5 cm × 16.6
cm × 13.5 cm heterogeneous peripherally enhancing lesion with multiple septations possibly arising
from right adrenal with subcentimetric abdominal and mesenteric lymphadenopathy and patient was referred to us. Hormonal evaluation including ONDST, serum
DHEA and urinary metanephrinand nor metanephrin were normal.
A provisional diagnosis of ACC was made and patient was taken
up for surgery. He underwent exploratory laparotomy via Thoracoabdominal
approach. No ascites or peritoneal or mesentric nodules
or lymphadenopathy. Liver surface had multiple 1x1 cm firm nodular
lesions in both lobes? hemangiomas. Tumor mass was seen arising
from right adrenal with multiple parasitic vessels, closely abutting
the IVC and pushing the right kidney inferiorly. Tumor was easily
separable from renal vessels and kidney. No invasion/ adhesions
with surrounding structures proceeded to right adrenalectomy. No
RPLND was done. Grossly (Figure 2) the tumor was well encapsulated.
Measuring 20 cm × 18 cm × 16 cm mass with variegated consistency,
weighing about 2 kg. Cut section showed heterogenous mass with
mixed solid cystic areas with areas of hemorrhage and necrosis.
Pale areas interspersed in between. Intraoperative and immediate
postoperative period was uneventful. Patient was discharged on sixth
postoperative day.
Final histopathology was inflammatory myofibroblastic tumor,
showing vague fascicles of benign spindle cells displaying plump
oval to elongated nuclei, dispersed chromatin and moderate amount
of cytoplasm associated with a moderate lymphoplasmacytic
inflammatory cell infiltrate, with focal areas of haemorrhage and
inconspicuous mitosis. Cystic areas lined by palisading fibroblasts
(Figure 3). Part of normal adrenal gland identified in the periphery.
Single lymph node identified was suggestive of reactive lymphoid
hyperplasia. IHC (Figure 4) was positive for Desmin, ALK-1 and
focal positivity for smooth muscle actin (SMA). CD 34 negative, Ki-
67 proliferation index was 3-4%.
Figure 1
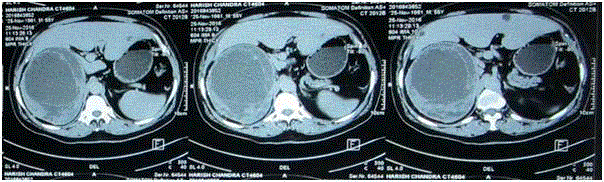
Figure 1
Heterogenous (17.5 cm × 16.6 cm × 13.5 cm) peripherally enhancing lesion with multiple septations arising from right adrenal with subcentimetric
abdominal and mesenteric lymphadenopathy. Lesion closely abutting upper pole of right kidney with no clear plane from right lobe of liver. High suspicion of
adrenocortical carcinoma. Opposite adrenal normal.
Discussion
In 1954, Umiker and Iverson coined the term “inflammatory
pseudotumor” because the clinical and imaging findings mimicked
those of malignant tumors [6]. IPT has been described by various names
in the literature: plasma cell granuloma (heart and lung), inflammatory
myofibroblastic tumor (lung), inflammatory myofibrohistiocytic
proliferation, histiocytoma, xanthoma, fibroxanthoma, fibrous
xanthoma, xanthogranuloma, xanthomatous pseudotumor, plasma
cell–histiocytoma complex (lung), plasmocytoma, solitary mast cell
granulomas, and inflammatory fibrosarcoma (urinary bladder) [7].
The exact cause of IPT is unknown. The confusion in nomenclature
is a result of ambiguity in determining the process of origin of these
tumors. There are two schools of thought: Post inflammatory reactive
process vs. true neoplasms. Post inflammatory theory is supported
by the fact that tumors are seen to occur post surgery, trauma and
VP shunts. Various organisms have been implicated in pathologic
specimens, including mycoplasmata and nocardiae in lung
pseudotumors, actinomycetes in liver, Epstein-Barr virus in splenic
and nodal pseudotumors, and mycobacteria in spindle cell tumors.
IL- 1contributes to the local and systemic effects. On the other hand
it has potential for local recurrence and distant metastases supporting
the neoplastic theory. Most definitive evidence of neoplastic origin
rests on the frequently seen clonal alterations involving the anaplastic
lymphoma kinase (ALK) gene located at chromosome 2p23 in a
significant subset of cases [8]. The term ‘inflammatory myofibroblastic
tumour’ (IMT) was introduced in 1986 to describe lesions previously
known as plasma cell granuloma or inflammatory pseudotumour
[9]. IMTs are uncommon tumours of spindle-cell proliferation with
predominance of myofibroblasts and histiocytes that may involve any
site abdomen, lungs, orbit, gastrointestinal and genitourinary tracts.
Most commonly seen in the lungs of children and adolescents, but it
can occur in older persons [2,7].
IMTs are usually asymptomatic, although 10% to 20% of cases are
associated with pyrexia and weight loss. Tumour size at diagnosis is
most often in the range of 5 cm to 10 cm. The biological behavior is
indeterminate. Clinical differentials include adrenocortical tumours,
pheochromocytoma, adrenal metastases and tuberculosis [1].
Imaging features are generally nonspecific and preoperative
differentiation from other adrenal masses is very difficult. The
radiologic features of IMT are variable and nonspecific possibly
because of the amount of fibrosis and cellular infiltration. On
ultrasound images, lesions can be hypoechoic or hyperechoic with
ill-defined or well-circumscribed borders with increased vascularity
during Color Doppler examinations. CECT shows varying
appearances with lesions showing low, equal, or high attenuation
compared with the surrounding tissues. Calcification can occur
and is more common in children. Post contrast images show a
variety of patterns with early peripheral and delayed central filling
or heterogenous, homogeneous and no enhancement at all. Larger
lesions may have central necrosis [10]. On MRI, IMTs usually have
low signal intensity on both T1- and T2-weighted images, which may
reflect the fibrotic nature of these lesions [11]. IPT are FDG-avid on
PET/CT images. PET/CT is highly sensitive but has low specificity
for IPT [12]. Complete surgical resection is mandatory as malignancy
cannot be excluded preoperatively [1]. The diagnosis of IMT is almost
always a histological surprise, as seen in our case. Surgical resection is
recommended even in cases with recurrence [11]. There is no role of
Radiation or Chemotherapy described for adrenal tumors. Radiation
and corticosteroids have been used to treat patients of Lung IMTs
who cannot undergo surgery. However there is no proven survival
benefit compared with surgery. With radical excision, the risk of
relapse is low, but relapse and distal metastasis occur many years after
treatment [12].
Microscopy shows circumscribed tumour composed of plump,
spindle cells or myofibroblasts and histiocytoid cells arranged
haphazardly or in fascicles with intervening thick collagen bundles,
accompanied by prominent infiltrate of chronic inflammatory cells,
particularly plasma cells (best distinguishes IMT from fibromatosis and
fasciitis). The advent of immunohistochemistry brought increasing
awareness of its distinctive features. IMTs usually express actin and
also desmin and keratin. Our case was positive for desmin, ALK and
SMA. ALK gene rearrangements, resulting in over expression of ALK
protein, are present in 30% to 40% of cases, mainly seen in pediatric age group. IMTs that have metastasized are most often ALK negative
[2,15] Hence ALK positivity can be considered a good prognostic
factor. No reported recurrences fo adrenal IMTs [1]. Other sites,
10% to 25% of patients have local recurrence. Less than 5% of cases
metastasize, but their behavior is difficult to predict on morphological
grounds, and it is increasingly believed that all cases of IMT are best
regarded as low-grade sarcomas. Sarcomatous degeneration has been
reported in IMTs at other sites, hence surveillance is crucial.
Figure 2
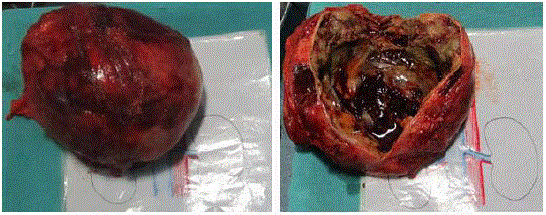
Figure 2
(A) Grossly the tumor was well encapsulated. Measuring 20 cm ×
18 cm × 16 cm mass, variegated in consistency, weighing about 2 kg. (B) Cut
section showed heterogenous mass with mixed solid cystic areas with areas
of hemorrhage and necrosis. Pale areas interspersed in between.
Figure3
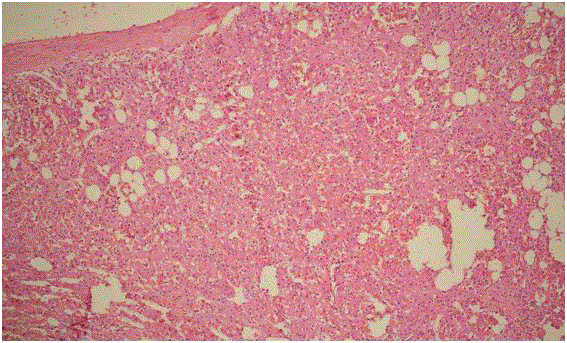
Figure 3
Histopathology showed (H&E stain, 10x) vague fascicles of benign
spindle cells with plump oval to elongated nuclei, dispersed chromatin,
moderate cytoplasm and lymphoplasmacytic inflammatory infiltrate with focal
areas of haemorrhage and inconspicuous mitosis. Part of normal adrenal
gland identified in the periphery.
Figure 4
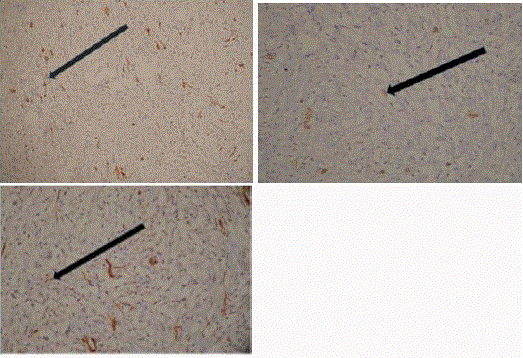
Figure 4
Immunohistochemistry strongly positive for (A) Desmin, (B) ALK-1
and (C) focal positivity for SMA.
Conclusion
IMTs of the adrenal are rare. Clinically and radiologically they mimic a malignant process and must be considered in the differential of radiologically suspicious adrenal masses. Complete surgical resection is mandatory as malignancy cannot be excluded preoperatively. Recurrence or Metastasis of adrenal IMTs is unknown but in view of metastatic potential of lesions at other sites, careful postoperative follow up including repeat imaging at 6 months to exclude local recurrence is advised.
References
- Tran-Dang MA, Banga N, Khoo B, Bates AW. Inflammatory myofibroblastic tumour arising in the adrenal gland: a case report. J Med Case Rep. 2014;8:411.
- Pettinato G, Manivel JC, De Rosa N, Dehner LP. Inflammatory myofibroblastic tumor (plasma cell granuloma). Clinicopathologic study of 20 cases with immunohistochemical and ultrastructural observations. Am J Clin Pathol. 1990;94(5):538-46.
- Luo LK, Shen HF, Zhou SY, Li JM, Xu WX. Inflammatory myofibroblastic tumour of adrenal. Zhonghua Bing Li Xue Za Zhi. 2006;35(4):252-3.
- Wang T-Y, Chou J-W, Shih Y-S. Inflammatory myofibroblastic tumour mimicking adrenal incidentaloma. Inter Med. 2011;50(2):165-6.
- Chawla A, Hameed Z, Mishra D, Monappa V. Adrenal inflammatory myofibroblastic tumour. BMJ Case Rep. 2013;2013.
- Umiker WO, Iverson LC. Post inflammatory tumor of the lung: report of four cases simulating xanthoma, fibroma or plasma cell granuloma. J Thorac Surg. 1954;28:55-62.
- Patnana M, Sevrukov AB, Elsayes KM, Viswanathan C, Lubner M, Menias CO. Inflammatory Pseudotumor: The Great Mimicker. AJR Am J Roentgenol. 2012;198(3):W217-W227.
- Mali VP, Tan HC, Loh D, Prabhakaran K. Inflammatory tumor of Retroperitoneum: A case report. Ann Acad Med Singapore. 2005;34(10):632-5.
- Day DL, Sane S, Dehner LP. Inflammatory pseudotumor of the mesentery and small intestine. Pediatr Radiol. 1986;6(3):210-5.
- Lim JH, Lee JH. Inflammatory pseudotumor of the liver: ultrasound and CT features. Clin Imaging. 1995;19(1):43-6.
- Narla LD, Newman B, Spottswood SS, Narla S, Rajasekhar K. Inflammatory Pseudotumor. Radio Graphics. 2003;23(3):719-29.
- Huellner MW, Schwizer B, Burger I, Fengels I, Schläpfer R, Bussmann C, et al. Inflammatory pseudotumor of the lung with high FDG uptake. Clin Nucl Med. 2010;35:722-3.
- Melloni G, Carretta A, Ciriaco P, Arrigoni G, Fieschi S, Rizzo N, et al. Inflammatory pseudotumor of the lung in adults. Ann Thorac Surg. 2005;79:426-32.
- Coffin CM, Hornick JL, Fletcher CD. Inflammatory myofibroblastic tumour; comparison of clinicopathologic, histologic, immunohistochemical features and ALK expression in atypical and aggressive cases. Am J Surg Pathol. 2007;31(4):509-20.
- Marino-Enriquez A, Wang WL, Roy A, Lopez-Terrada D, Lazar AJ, Fletcher CD, et al. Epithelioid inflammatory myofibroblastic sarcoma; an aggressive intra-abdominal variant of inflammatory myofibroblastic tumour with nuclear membrane or perinuclear ALK. Am J Surg Pathol. 2011;35(1):135-144.