Review Article
Use of Omega-3s in Traumatic Brain Injury
Michael D Lewis*
Brain Health Education and Research Institute, USA
*Corresponding author: Michael D Lewis, Brain Health Education and Research Institute, 7811 Montrose Road #215, Potomac, MD 20854, USA
Published: 28 Apr, 2017
Cite this article as: Lewis MD. Use of Omega-3s in
Traumatic Brain Injury. Clin Surg. 2017;
2: 1449.
Abstract
Clinical studies thus far have failed to identify an effective treatment strategy for Traumatic
brain injury (TBI), when a combination of targets controlling aspects of neuroprotection,
neuroinflammation, and neuroregeneration is needed. Omega-3 fatty acids (n-3FA) offer the
advantage of this approach. Although further clinical trial research is needed, there is a growing
body of strong preclinical evidence and clinical experience which suggests that benefits may be
possible from aggressively adding substantial amounts of n-3FA to optimize the nutritional
foundation of TBI, concussion, and post-concussion syndrome patients. Early and optimal doses
of n-3FA, even in a preventive setting, have the potential to improve outcomes without significant
side-effects. With evidence of unsurpassed safety and tolerability, n-3FA should be considered in
the acute care and surgical setting, if conventional medicine can overcome its inherent bias against
nutritional, non-pharmacologic therapies.
Keywords: Omega-3; Decosahexanoic acid; DHA; TBI; Brain injury; Concussion;
Neuroregeneration; Neuroinflammation; Neuroprotection
Introduction
Omega-3 polyunsaturated fatty acids (n-3FA) are structural components of cell membranes,
particularly docosahexaenoic acid (DHA) which is most concentrated in the brain and retina.
Emerging science on the ability of n-3FA to be beneficial to the nervous system during and after
acute traumatic brain injury (TBI) is acknowledged, mainly in preclinical studies, but now in
growing clinical experience and case reports.
TBI has long been recognized as a leading cause of traumatic death and disability. TBI is caused
by a bump, blow or jolt to the head or a penetrating head injury that disrupts the normal function
of the brain. Over 3.5 million known TBI’s occur annually, approximately 52,000 deaths, and more
than 300,000 hospitalizations in the United States alone [1]. TBI is often classified using mild,
moderate, and severe categories. It is believed that 80% to 95% of all TBI are mild, often labeled as a
“concussion” and are not seen in hospital settings [1]. TBI, most often from falls, vehicle accidents,
contact sports, and violence, is a major healthcare concern, constituting a major cause of death and
disability not just in the United States, but throughout the world. Motor bikes are major causes,
increasing in significance in developing countries as other causes reduce. Some consider TBI a
global public health epidemic [2].
Classically, TBI is described as occurring in two phases, or on the basis of the pathophysiologic
mechanism. The primary or initial injury occurs as a direct result of the traumatic event itself. A
secondary injury, or phase, occurs from multiple neuropathologic processes that can continue for
days to weeks following the initial insult. The primary injury is immediate and not amenable to
treatment, only prevention. If severe enough, death can occur almost instantaneously. The damage
that occurs from the primary injury is complete by the time medical care can be instituted. Highspeed
collisions with very rapid deceleration are particularly injurious, but sports-related injuries
also can be devastating. Because the neuronal structures reside in a fluid-filled compartment, they
often lag behind the bony structure as it moves during the sudden stopping of the body in motion.
The brain often strikes both in the direct and opposite plane of motion against the inner bony table.
This is the coup-contre-coup pattern, where contusions to the brain are seen at the site of skull
impact and 180 degrees opposite the site of impact [3].
The secondary injury of TBI is a prolonged pathogenic process leading to cell death and
worsening damage to the brain far beyond the primary injury (Figure 1). The secondary injury
phase of TBI consists of: ischemia, excitotoxicity, and intracellular biochemical cascades; axonal
injury; cerebral edema; and inflammation and regeneration. The importance of the secondary injury
has gained widespread recognition as a potential target of therapeutic intervention. Although much
has been learned about the molecular and cellular mechanisms of TBI
in the past two decades, these advances have failed to translate into a
successful clinical trial and no significant improvement in treatment
beyond the acute setting [3].
Neuroinflammation is complicated, beyond the scope of this
review. A critical balance exists between repair and pro-inflammatory
factors that determine the outcome of neurodegenerative processes.
Acute inflammation in the brain is characterized by rapid activation of
the innate immune cells of the central nervous system, microglia and
astrocytes [4]. Once activated, astrocytes, the most abundant cells in
the brain, release various growth factors, cytokines, and chemokines
that function as neuromodulators to regulate inflammation. Common
cytokines produced in response to brain injury include: interleukin-6
(IL-6), which is produced during astrogliosis, and interleukin-1 beta
(IL-1β) and tumor necrosis factor alpha (TNF-α), which can induce
neuronal cytotoxicity [5]. Overturning the long-held concept of the
absence of lymphatic vasculature in the central nervous system, it
is now known that the brain is directly connected to the peripheral
immune system via the "glymphatic" pathway changing entirely the
way the neuro-immune interaction is perceived [6].
The concept that TBI can lead to neurodegenerative changes was
first introduced in 1926 by neurologists Osnato and Giliberti [7].
Affected individuals often exhibit disordered memory and executive
functioning and behavioral and personality disturbances (e.g.,
apathy, depression, irritability, impulsiveness, and suicidality). Upon
autopsy, the presence of hyperphosphorylated tau protein deposition,
whether it be in the form of neurofibrillary tangles (NFTs), neuropil
threads (NTs), or glial tangles (GTs), is a defining feature of Chronic
Traumatic Encephalopathy (CTE) [8]. CTE in a retired professional
American foot ball player was first recognized in 2002 by Omalu et al.
[9] when autopsying the brain of a deceased player.
Nearly lost in the discussions of CTE has been the role of sustained
or chronic neuroinflammation, even though this association has been
well established pathologically since the 1950s. It has been widely
believed that the accumulation of toxins and pathological proteins
were an issue of over production rather than poor clearance from the
brain. The recent discovery of the glymphatic pathway facilitating the
clearance of β-amyloid and tau from the brain may overturn that belief
[6]. After TBI however, glymphatic pathway function was reduced by
~60%, with this impairment persisting for at least 1 month post injury.
Such chronic impairment of glymphatic pathway function after TBI
may be a key factor that renders the post-traumatic brain vulnerable
to tau aggregation and the onset of neurodegeneration [10].
Figure 1
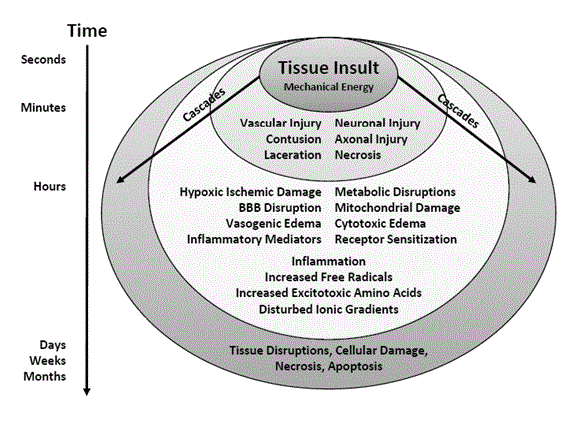
Figure 1
The primary injury of TBI is caused by a transfer of mechanical injury to the brain tissue. This is followed by the secondary injury that occurs over minutes
to hours to days and even weeks and months. It is characterized by numerous metabolic and biochemical cascades that may cause more damage than the initial
tissue insult itself.
Figure 2
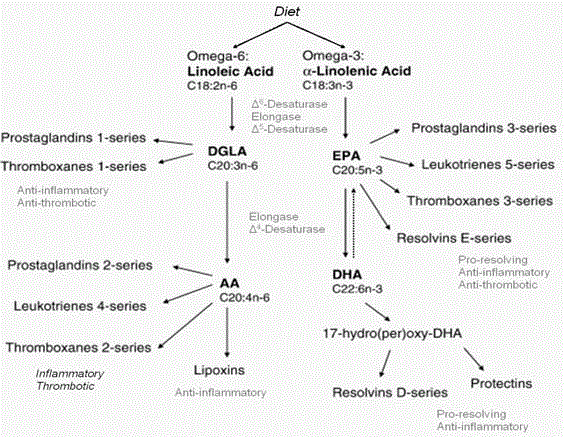
Figure 2
Omega-6s and omega-3s compete for the same elongation and desaturation enzymes. However, very little ALA is converted ultimately to DHA mainly
because the amount of omega-6s in the typical Western diet overwhelms the pathways favoring the synthesis of arachidonic acid. Note the pro-inflammatory and
anti-inflammatory mediators.
Figure 3
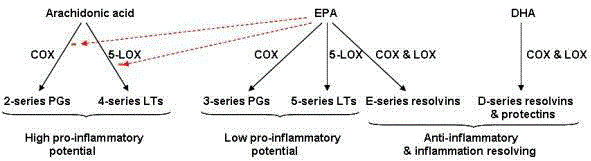
Figure 3
The downstream biochemical pathways of AA, EPA, and DHA result in stimulation of pro-inflammatory 2-series prostaglandins and 4-series leukotrienes;
anti-inflammatory 3-series prostaglandins and 5-series leukotrienes; E- and D-series resolvins; and neuroprotectins [20].
Failure to Find Therapeutic Interventions for TBI
The most definitive strategy to avoid short- or long-term
detrimental effects of all TBI, mild to severe, is through primary
prevention or avoidance of the injury in the first place. However,
once a TBI occurs, the secondary injury represents a window of
opportunity for therapeutic intervention with the potential to
prevent and/or reduce brain damage and improve long-term patient
outcome. To date, however, promising preclinical results have not
been translated into successful clinical trials [11]. This may be due to
the fact that most interventions target a single biochemical cascade
rather than multiple mechanisms of injury.
Approaches that target multiple aspects of TBI are needed.
The Western medical system evolved around the epidemiological
triad of acute infectious diseases: one host-agent-environment and
subsequently one drug to cure. Pharmaceuticals by nature are aimed
at disrupting single enzymatic processes. TBI is too complicated
for such a narrow approach. What is needed is a broad spectrum,
more holistic approach. Interventions targeting all aspects of the
secondary injury, plus repair, regeneration, and protection of the
brain are desperately needed. Effective interventions should also treat
persistent symptoms associated with the long-term effects of TBI
(post-concussive symptoms, e.g., memory disturbances, depression,
headache) [12].
The Role of Omega-3 Fatty Acids in the Brain
It is well recognized that n-3FA are important for proper
neurodevelopment and function. Linoleic acid (a short-chain n-6FA)
and Alpha-Linolenic Acid (ALA; a short-chain n-3FA) are fatty acids
that cannot be made de novo, must be consumed in the diet, and are
therefore considered essential. They are precursors for the synthesis
of longer, more bioactive poly Unsaturated Fatty Acids (PUFAs)
such as the n-6FA, Arachidonic Acid (AA), and the n-3FAs, Eicosa
Pentaenoic Acid (EPA) and DHA. However, n-6FA and n-3FA
compete for the same elongation and desaturation enzymes and the
conversion of ALA to EPA and DHA in humans is negligible (Figure
2). Therefore EPA, and DHA in particular, should be consumed
directly in the diet [13].
The age-old saying, “You are what you eat,” holds true here.
The composition of neuronal cell membranes is directly reflected
by the dietary intake of n-3FA and n-6FA. The ratio of n-6 and
n-3 FAs affects the physiological functions of the brain, changes in
cell permeability and synaptic membrane fluidity, and has a major
influence on the activity of neurotransmitters [14]. Unfortunately,
today’s Western dietary intakes result in an over dominant intake
of pro inflammatory n-6FA creating a relative deficiency of immune
modulating n- 3FA. The evolutionary human diet, up until the last
century, had a relatively even AA: DHA ratio of approximately 1:1,
was high in fiber, rich in fruits, vegetables, lean meat, and fish, thus
provided a more balanced ratio between n-6FA and n-3FA [14]. That
ratio is now approximately 22-25:1 with n-6FA dominating [15]. The
estimated per capita consumption of soybean oil, the most common
source of n-6FA in the Western diet, increased greater than a 1000-
fold from 1909 to 1999 in the United States, now contributing almost
8% of all calories consumed [16]. Excessive consumption of AA
displaces DHA from membrane phospholipids reflected directly in
the composition of neuron membrane phospholipids overwhelmingly
favoring AA-derived inflammatory processes [17].
AA, the primary n-6FA in the brain, is metabolized by
cyclooxygenase and lipoxygenase enzymes to proinflammatory
eicosanoids. Eicosanoids are key mediators and regulators of
inflammation involved in modulating the intensity and duration of
inflammatory responses. AA is the major precursor for eicosanoid
mediators such as two-series prostaglandins and thromboxanes,
prostaglandin E2 (PGE2); and leukotriene B4 (LTB4). These
eicosanoids enhance vascular permeability, increase local blood flow,
increase infiltration of leukocytes, and enhance production of other
proinflammatory cytokines [18].
In contrast, n-3FA, are anti-inflammatory. EPA is also a substrate
for the cyclooxygenase and lipoxygenase enzymes that produce
eicosanoids, but the mediators produced are biologically different
from the AA-derived mediators. EPA-derived eicosanoids antagonize
the action of eicosanoids derived from AA, thus can decrease AAderived
cyclooxygenase activity and inhibit the formation of these
proinflammatory eicosanoids and cytokines [19]. EPA and DHA also
give rise to E-series and D-series resolvins and protectins (Figure 3). Eand
D-series resolvins decrease accumulation of polymorphonuclear
leukocytes (PMNs) and attenuate pro-inflammatory signaling.
Resolution of inflammation via E- and D-series resolvins is required
to shut off ongoing inflammatory processes and limit tissue damage.
The anti-inflammatory effects of n-3FA suggest a therapeutic value, at
the least, the opportunity, to modulate the inflammatory aspect of the
secondary injury phase of TBI [20].
While EPA is well known for its beneficial vascular properties,
very little is found in brain tissue. DHA, on the other hand, is
highly concentrated in the central nervous system and is essential
for proper neuronal and retinal function. DHA is present in
high concentrations in neurons where it is esterified to neuronal
cell membrane phospholipids in phosphatidylserine (PS) and
phosphatidylethanolamine (PE) [21]. DHA signalolipidomics is
directly affected by the dietary supply of DHA. When mobilized from
the cell membrane by phospholipase A2 (PLA2), DHA regulates unique
cellular and molecular signaling pathways. Whereas eicosanoids are
derived from 20-carbon chain AA and EPA, docosanoids, including
neuroprotectin D1 (NPD1) proteins, are derived from the 22-carbon
DHA in response to cellular stress and elicit neuroprotection [22].
DHA also promotes neurite growth, increased neurite branching, and
subsequent synaptogenesis, resulting in enhanced synaptic function
and improving neuronal repair after injury [23].
Omega-3 PUFAs and TBI
EPA and DHA have the ability to impact all the main mechanisms
of the secondary injury phase of TBI; have neuroregenerative
properties; is well-studied as substances in the scientific literature;
can be given to a patient during the acute phase of injury (or prior to
injury) and continued throughout the patient’s entire rehabilitation;
and can be used prophylactically prior to injury in populations at risk
of TBI. Animal models of neurologic pathology indicate n-3FA have
the potential for improvement in the outcomes of TBI [24,25], stroke
model [26], and spinal cord injury (SCI) [27].
Three case studies are now reported in the scientific literature
that can provide clinical guidance. In January 2006, an explosion
in the Sago Mine in central West Virginia resulted in 14 trapped
miners. Forty hours later, one lone survivor was found and brought
to medical care. He had suffered hypoxia and exposure to toxic gases,
dehydration, and rhabdomyolysis. The patient demonstrated many
classic features of carbon monoxide toxicity, including neurologic,
cardiac, and renal dysfunction as well as respiratory failure. In
addition to rapid resuscitation, dialysis, and hyperbaric oxygen
therapy, starting on hospital day 8, the patient was treated with 21.2
g per day of n-3FA that contributed to his neurological recovery
following an initial presentation in deep coma. On day 21, he was
transferred to a rehabilitation facility and discharged to home two
months later [28].
In March 2010, a teenager sustained a severe TBI with diffuse axonal
injury from a motor vehicle accident. The attending neurosurgeon's
impression was that the injury was likely lethal. Believed to be in
a permanent vegetative state, a tracheotomy and percutaneous
endoscopic gastrostomy (PEG) tube were placed for custodial care
and enteral feedings were started on day 10. The next day, the patient
began receiving almost 20 g total n-3FA daily via his PEG based on
the clinical results of the Sago Mine survivor’s experience. On day
21, he was weaned off the ventilator and transported to a specialized
rehabilitation institute. He was discharged to home four months after
the injury [29].
When this case was reported on CNN’s Sanjay Gupta MD show,
the mother of an 8-year-old girl requested her comatose daughter be
given high dose fish oil 82 days following a near drowning accident
in August 2012. The patient had sustained severe anoxic brain injury
in the setting of prolonged cardiac and respiratory arrest. The patient
was discharged to home one month later [30]. In none of these three
case reports, were any side effects noted. In spite of these case reports
and anecdotal evidence, there is a complete lack of clinical trials for
the use of n-3FA in human TBI.
Suggested Protocols for Using Omega-3s for TBI
Because of the complex heterogeneity of TBI and without
definitive clinical trial evidence, there is no way to know if n-3FA
will help in any particular case of TBI. Growing clinical experience by
numerous providers is that the brain needs to be saturated with high
doses of n-3FA in order for the brain to have the opportunity to heal.
Without an optimal supply of omegas, healing is less likely to happen.
It is well recognized that n-3FA are not a drug and not a cure and
every situation is different. Clinically, some patients respond better
than others. However, there is no downside to providing optimal
levels of nutrition in order to give a patient the best opportunity to
regain as much function as possible following a TBI.
The dose that was used for the March 2010 case [29] was: a
concentrated liquid, one tablespoon (15 ml) twice a day for a total of
30 ml per day in the feeding tube followed by a saline flush providing
9,756 mg EPA, 6,756 mg DHA, and 19,212 mg total n-3FA daily.
This case received this dose for about a year without any problems
or side effects. While these doses were used in adults, in pediatric
patients, lower doses should be considered. Most importantly, weekly
analysis of the AA/EPA ratio should be monitored to determine the
appropriate dosage to avoid theoretical possibility of bleeding. When
that ratio drops below 1.0, the dosage should be decreased as needed
and intermittent monitoring continued [30].
For concussions and milder cases of TBI when patients are able
to swallow on their own, a different protocol with fish oil capsules
or liquid equivalent has been used extensively as published by the
nonprofit charity Brain Health Education and Research Institute on
their website (http://www.brainhealtheducation.org): concentrated
fish oil in triglyceride form providing approximately 3000 mg of
n-3FA per dose is consumed three times a day for a minimum of
one week before decreasing to twice a day and eventually once a day.
Anecdotally, this loading dose approach provides a more immediate
benefit improving mood, calmness, headaches, and cognitive
function and the large doses in the beginning act to overcome
the relative deficit of n-3FA in most people. One open label study
evaluating pre- and post therapy supplementation for five weeks using
electroencephalogram (EEG) brain mapping has been completed
and submitted for publication. The pilot study showed statistically
significant improvement in patient brain auditory evoked response
processing and heart rate variability power and improvements in
most EEG, evoked response, and heart rate variability variables in
five of seven patients.
Potential Harmful Effects
Due to the known anti-thrombotic action of these compounds, it is commonly believed they may increase the risk of excessive bleeding or even hemorrhagic stroke. Theoretically, the biochemistry of n-3FAs tells us this should be true. However, that has never been shown to be of clinical concern in any clinical trial reported in the literature. In fact, the anti-thrombotic nature is one of the properties that make n-3FA effective in decreasing mortality, particularly cardiovascular mortality where the effect is more beneficial than statins [31]. Multiple clinical trials have shown that high dose fish oil consumption is safe, even in patients receiving other agents that may increase the risk of bleeding, such as aspirin and warfarin [32]. Interestingly, it is standard of care that most critically ill and injured patients are put on subcutaneous heparin, or similar, to prevent deep vein thrombosis while immobile. Recently, Farooqui et al. [33], examined the use of blood thinning pharmaceuticals and concluded they are safe, do not increase the risk of intracranial hemorrhage, and decreases the rate of deep vein thrombosis and pulmonary embolism. Potent blood thinners used in this protocol (heparin and Lovenox) completely block the enzymes responsible for allowing the platelets to clot. n-3FA potentiate the body’s natural anti-clotting abilities rather than blocking enzymatic processes and add the ability to modulate neuroinflammation, decrease apoptosis, and start synaptogenesis. Ironically, most doctors who will not use n-3FA citing that high doses of n-3FA decrease the ability of blood to clot and increase a patient’s risk of bleeding, immediately put their ICU patients on potent pharmaceutical blood thinners that increase the risk far greater than that of n-3FA.
Conclusions and Future Directions
TBI, with their diverse heterogeneity and prolonged secondary
pathogenesis, remain a clinical challenge to clinician, patients,
and their families. Current medical management of TBI patients
appropriately focuses on specialized prehospital care, intensive acute
clinical care, and long-term rehabilitation, but lacks clinically proven
effective management with neuroprotective and neuroregenerative
agents [11]. Clinical studies thus far have failed to identify an effective
treatment strategy as they typically have targeted single enzymatic
factors in an attempt to identify a pharmacologic target rather than
considering multiple mechanisms of injury with a more holistic
approach. The concept of a ‘magic bullet’ focused on a single target is
not helpful, and instead a combination of targets controlling aspects
of neuroprotection, neuroinflammation, and regeneration is needed.
n-3FA offer the advantage of this poly-target approach [27].
Although further clinical trial research is needed to establish the
true advantage of using n-3FA, there is a growing body of strong
preclinical evidence and clinical experience suggests that benefits
may be possible from aggressively adding substantial amounts
of n-3FA to optimize the nutritional foundation of TBI patients.
Numerous university athletic programs and professional sports
teams are reported to be using n-3FA and the omega protocols with
their athletes for the prevention of brain injuries. However, recovery
from TBI may be hindered by our modern, pro-inflammatory diet.
An optimal nutritional regimen to overcome the n-6FA dominance
must be in place if the brain is to be given the best opportunity to
repair itself.
Administration of substantial and optimal doses of n-3FA earlier
in the course of TBI, even prophylactically in those at greater risk of a
brain injury, such as soldiers and athletes, has the potential to improve
outcomes from this potentially devastating public health problem
[34]. As the father of one severe TBI survivor says, “Conventional
medicine only takes survivors of severe TBI so far, often ending at the
nursing home door, or heavily medicated at home, facing long empty
hours, and overwhelming family resources. Unconventional therapies
are not merely a reasonable option, they are a necessity” [20]. With
evidence of unsurpassed safety and tolerability, n-3FA should be
considered mainstream, conventional medicine, if conventional
medicine can overcome its inherent bias against nutritional, nonpharmacologic
therapies.
References
- Coronado VG, McGuire LC, Sarmiento K, Bell J, Lionbarger MR, Jones CD, et al. Trends in Traumatic Brain Injury in the U.S. and the public health response: 1995-2009. J Safety Res. 2012;43(4):299-307.
- Rodríguez-Rodríguez A, Egea-Guerrero JJ, Murillo-Cabezas F, CarrilloVico A. Oxidative stress in traumatic brain injury. Curr Med Chem. 2014;21(10):1201-11.
- Ling GS, Marshall SA. Management of traumatic brain injury in the intensive care unit. Neurol Clin. 2008;26(2):409-26.
- Streit WJ, Mrak RE, Griffin WS. Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation. 2004;1(1):14.
- Ramesh G, MacLean AG, Philipp MT. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediators Inflamm. 2013;2013:480739.
- Louveau A, Smirnov I, Keyes T, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523(7560):337-41.
- Ling H, Hardy J, Zetterberg H. Neurological consequences of traumatic brain injuries in sports. Mol Cell Neurosci. 2015;66:114-22.
- Gavett BE, Stern RA, McKee AC. Chronic traumatic encephalopathy: a potential late effect of sport-related concussive and subconcussive head trauma. Clin Sports Med. 2011;30(1):179-88.
- Omalu B, DeKosky S, Minster R, Kamboh M, Hamilton R, Wecht CH. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery. 2005; 57(1):128-34.
- Iliff J, Chen M, Plog B, Zeppenfeld D, Soltero M, Yang L, et al. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J Neurosci. 2014;34(49):16180-93.
- Xiong Y, Mahmood A, Chopp M. Emerging treatments for traumatic brain injury. Expert Opin Emerg Drugs. 2009;14(1):67-84.
- Diaz-Arrastia R, Kochanek P, Bergold P, Kenney K, Marx CE, Grimes JB, et al. Pharmacotherapy of traumatic brain injury: state of the science and the road forward report of the Department of Defense Neurotrauma Pharmacology Workgroup. J Neurotrauma 2014;31(2):135-58.
- Salem N Jr, Litman B, Kim HY, Gawrisch K. Mechanisms of action of docosahexaenoic acid in the nervous system. Lipids. 2001;36(9):945-59.
- Farooqui AA. n-3 fatty acid-derived lipid mediators in the brain: new weapons against oxidative stress and inflammation. Curr Med Chem. 2012;19(4):532-43.
- Lewis MD, Hibbeln JR, Johnson JE, Lin YH, Hyun DY, Loewke JD. Suicide deaths of active-duty US military and omega-3 fatty-acid status: a casecontrol comparison. J Clin Psychiatry. 2011;72(12):1585-90.
- Blasbalg TL, Hibbeln JR, Ramsden CE, Majchrzak SF, Rawlings RR. Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century. Am J Clin Nutr. 2011;93(5):950-62.
- Bradbury J. Docosahexaenoic acid (DHA): an ancient nutrient for the modern human brain. Nutrients. 2011;3(5):529-54.
- Bailes JE, Mills JD. Docosahexaenoic acid reduces traumatic axonal injury in a rodent head injury model. J Neurotrauma. 2010;27(9):1617-24.
- Lewis MD, Bailes J. Neuroprotection for the warrior: dietary supplementation with omega-3 fatty acids. Mil Med. 2011;176(10):1120-7.
- Calder PC. Omega-3 fatty acids and inflammatory processes. Nutrients. 2010;2(3):355-74.
- Kim HY. Biochemical and biological functions of docosahexaenoic acid in the nervous system: modulation by ethanol. Chem Phys Lipids. 2008;153(1):34-46.
- Bazan N, Molina M, Gordon W. Docosahexaenoic acid signalolipidomics in nutrition: significance in aging, neuroinflammation, macular degeneration, Alzheimer’s, and other neurodegenerative diseases. Ann Rev Nutr.2011;31:321-51.
- Kim HY, Spector A. Synaptamide, endocannabinoid-like derivative of docosahexaenoic acid with cannabinoid-independent function. Prostaglandins Leukot Essent Fat Acids.2013; 88(1):121-25.
- Wu A, Ying Z, Gomez-Pinilla F. Exercise facilitates the action of dietary DHA on functional recovery after brain trauma. Neuroscience. 2013;248:655-63.
- Mills JD, Hadley K, Bailes JE. Dietary supplementation with the omega-3 fatty acid docosahexaenoic acid in traumatic brain injury. Neurosurgery. 2011;68(2):474-81.
- Eady T, Khoutorova L, Obenaus A, Mohd-Yusof A, Bazan N, Belayev L. Docosahexaenoic acid complexed to albumin provides neuroprotection after experimental stroke in aged rats. Neurobiol Dis. 2014;62:1-7.
- Michael-Titus AT, Priestley JV. Omega-3 fatty acids and traumatic neurological injury: from neuroprotection to neuroplasticity? Trends Neurosci. 2014;37(1):30-8.
- Roberts L, Bailes J, Dedhia H, Zikos A, Singh A, McDowell D, et al. Surviving a mine explosion. J Am Coll Surg. 2008;207(2):276-83.
- Lewis M, Ghassemi P, Hibbeln J. Therapeutic use of omega-3 fatty acids in severe head trauma. Am J Emerg Med. 2013;31(1):273.
- Sears B, Bailes J, Asselin B. Therapeutic uses of high-dose omega-3 fatty acids to treat comatose patients with severe brain injury. Pharma Nutrition. 2013;1(3):86-9.
- Studer M, Briel M, Leimenstoll B, Glass TR, Bucher HC. Effect of different antilipidemic agents and diets on mortality: a systematic review. Arch Intern Med. 2005;165(7):725-30.
- Hasadsri L, Wang BH, Lee JV, Erdman JW, Llano DA, Barbey AK, et al. Omega-3 fatty acids as a putative treatment for traumatic brain injury. J Neurotrauma. 2013;30(11):897-906.
- Farooqui A, Hiser B, Barnes S, Litofsky N. Safety and efficacy of early thromboembolism chemoprophylaxis after intracranial hemorrhage from traumatic brain injury. J Neurosurg .2013;119(6):1576-82.
- Goldstein J. No Stone Unturned: A Father’s Memoir of His Son's Encounter with Traumatic Brain Injury. Virginia: Potomac Books, USA; 2012.