Case Report
Type a Aortic Dissection and Impaired Systolic Function in a Child with Loeys-Dietz Syndrome
Yan Li*, Qian Chang, Xiangyang Qian, Xiaogang Sun and Mingyao Luo
Department of Cardiovascular Surgery, National Center for Cardiovascular Diseases and Fuwai Hospital, China
*Corresponding author: Yan Li, Department of Cardiovascular Surgery, National Center for Cardiovascular Diseases and Fuwai Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, 167 Beilishi Rd, Beijing, 100037, China
Published: 06 Mar, 2017
Cite this article as: Li Y, Chang Q, Qian X, Sun X, Luo M.
Type a Aortic Dissection and Impaired
Systolic Function in a Child with Loeys-
Dietz Syndrome. Clin Surg. 2017; 2:
1329.
Abstract
Loeys-Dietz syndrome (LDS) is a recently recognized genetic aortic aneurysm syndrome
characterized by the triad of hypertelorism, bifid uvula or cleft palate, and generalized arterial
tortuosity with aneurysms and dissections throughout the arterial tree with a high risk of earlyonset
aortic dissection but with no known myocardial involvement [1]. We report a child who
was diagnosed as acute type A aortic dissection, a novel transforming growth factor beta (TGFβ)
receptor type I (TGFBR1) mutation, and impaired systolic function after urgent surgical repair.
Keywords: LDS: Loeys-dietz Syndrome; TGFβ: Transforming Growth Factor β; TGFBR1: TGF Beta-receptor Type I; LVEF: Left Ventricular Ejection Fraction
Case Presentation
A 7-year-old boy visited the emergency room with a complaint of sudden-onset chest pain and dyspnea. At the age of 6, this boy was diagnosed as aortic root aneurysm with mild aortic regurgitation. At the age of 7 years he showed height 140 cm and arm span 136 cm. A thorough physical examination demonstrated dolichocephaly, hypertelorism, hypoplastic alae nasi, higharched palate, microretrognathia, joint hypermobility, which suggested LDS. Echocardiography demonstrated a large aortic root aneurysm with a dissection membrane, left ventricle diameter 50 mm, and severe aortic regurgitation. Therefore, a CT imaging was made rapidly, which clarified acute type a aortic dissection without involvement of the coronary arteries and aortic root diameter 75 mm (Figure 1). Suspicion for LDS was confirmed by genetic testing, which identified a novel heterogeneous non-frame shift deletion c.G678_680del AGA in exon 4 of TGBFR1 in the highly conserved kinase domain of the gene, which could subsequently result in the heterogeneous p.Glu228del variation (Figure 2). An urgent operation was performed by a median sternotomy and total cardiopulmonary bypass (CPB). Aortic root replacement was done using a modified Bentall technique. The postoperative course was uneventful, and the patient was discharged 10 days postoperatively without any complications. The postoperative CT imaging and echocardiogram revealed patent epicardial coronary arteries, global systolic dysfunction (left ventricular ejection fraction, LVEF, 45%), and dilated left ventricle (left ventricle diameter 65 mm). He was treated with diuretics, digoxin, and an angiotensin-converting enzyme inhibitor. He developed fatigue on postoperative day 45, and echocardiogram revealed global systolic dysfunction again (LVEF 34%). His fatigue and left ventricular systolic function improved to 42% with medical therapy on postoperative day 90. Cardiomyopathy was suspected and a strict surveillance was recommended.
Figure 1
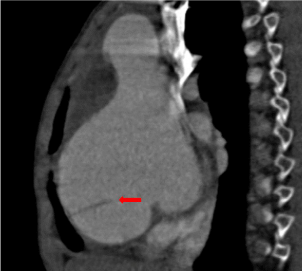
Figure 1
Sagittal reconstruction shows dilation of Valsalva sinuses with a dissection membrane (red arrow).
Figure 2
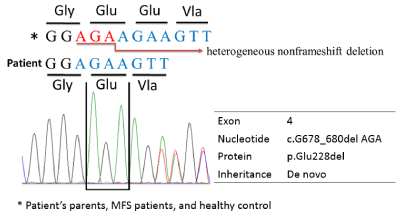
Figure 2
Sequence chromatogram of the patient, showing a novel
heterogeneous non-frameshift deletion c.G678_680del AGA in TGBFR1
exon 4, leading to p.Glu228del.
Discussion
The most important finding of LDS is the aggressive and rapid progression of aortic pathology even though the patients are young in age. Notably, what makes these TGFβ-associated cardiovascular diseases more challenging is that they affect many cardiac tissues in the same patients [3,4]. Only one case of cardiomyopathy due to microvascular dysplasia a LDS adult has been described [4], however, cardiomyopathy in LDS children has not previously been described to the best of our knowledge. Although it is possible that LDS and cardiomyopathy are unrelated, it seems plausible that the TGFBR1 mutation in LDS could cause microvascular dysplasia and subsequent myocardial dysfunction. Further study may provide an opportunity to clarify the significance of this finding.
References
- Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006; 355: 788-798.
- Eckman PM, Hsich E, Rodriguez ER, Gonzalez-Stawinski GV, Moran R, Taylor DO. Impaired systolic function in Loeys-Dietz syndrome: a novel cardiomyopathy? Circ Heart Fail. 2009; 2: 707-708.
- Das BB, Taylor AL, Yetman AT. Left ventricular diastolic dysfunction in children and young adults with Marfan syndrome. Pediatr Cardiol. 2006; 27: 256-258.