Review Article
How Wound Healing Abnormalities Impact Clinical Care
Stefanos Boukovalas1, Kristen A. Aliano1, Michael Smith J2 and Linda G. Phillips1*
1Department of Surgery, Division of Plastic Surgery, University of Texas Medical Branch, USA
2School of Medicine, University of Texas Medical Branch, USA
*Corresponding author: Linda G. Phillips, Department of Surgery, Division of Plastic Surgery, University of Texas Medical Branch, 301 University Blvd, Galveston, TX 77555-0724, USA
Published: 14 Oct, 2016
Cite this article as: Boukovalas S, Aliano KA, Michael
Smith J, Phillips GL. How Wound
Healing Abnormalities Impact Clinical
Care. Clin Surg. 2016; 1: 1158.
Abstract
One of the greatest clinical challenges faced by plastic surgeons is that of the chronic non-healing wound. In this review, we provide a brief overview of the molecular biology of wound healing,
discussing the three overlapping phases: reactive, proliferative and maturational. We also discuss
the most commonly encountered deterrents to physiologic wound healing, including hypoxia,
infection, diabetes, malnutrition, and medications. Moreover, we describe the most common
chronic wounds: pressure ulcers, diabetic foot ulcers, and venous stasis wounds. A variety of
treatment modalities is available to solve these difficult problems, such as hyperbaric oxygen therapy
and regenerative medicine treatments. Due to the complex natural history of chronic wounds, they
are best approached in an inter-disciplinary manner.
Keywords: Wound healing; Diabetic foot ulcers; Inflammation
Introduction
Chronic, non-healing wounds pose a formidable clinical challenge for plastic and reconstructive surgeons. A thorough understanding of the underlying physiology of wound healing is required to optimize patient outcomes and understand the complex and ever-expanding armamentarium of therapeutic options used to treat chronic wounds. This review provides a brief overview of the molecular biology of wound healing, deterrents to physiologic wound healing, types of chronic wounds commonly encountered by plastic surgeons, current treatment modalities, and future therapeutic horizons for treating patients with chronic wounds.
Normal Wound Healing
Wound healing is a complex process, which is classically divided into three overlapping phases:
reactive, proliferative, and maturational (Figure 1,2) [1].
I. Reactive phase
A) Hemostasis: Following tissue injury, exposed subendothelial collagen activates platelets
and the intrinsic clotting cascade. Activated platelets adhere to one another to form a hemostatic
plug, activate the extrinsic clotting cascade, and release a myriad of soluble factors, which promote
coagulation and inflammation.
B) Inflammation: Leukocytes are recruited to the area of injury by soluble mediators released
from activated platelets and endothelial cells. Neutrophils are the first cells to arrive in the wound,
often appearing within 24-48 hours of the time of injury. Neutrophils enter the wound site through
diapedesis and subsequently work to clear necrotic debris, foreign material, and bacteria through the
generation of reactive oxygen species. Subsequently, macrophages enter the wound at roughly 48-
96 hours post-injury to phagocytose bacteria, digest extracellular debris, and secrete several soluble
mediators, which coordinate later stages of the wound-healing cascade including angiogenesis,
fibroblast activation/proliferation, and collagen production/maturation. Lymphocytes are the last
class of leukocytes to enter the wound site, often arriving within 5-7 days of injury. Lymphocytes
primarily function to sustain the inflammatory response through the release of cytokines and
promotion of long-term immunity to pathogens encountered in the wound.
II. Proliferation
The proliferative phase of wound healing results in granulation tissue formation. In response
to hypoxia, epithelial cells release vascular endothelial growth factor (VEGF) which stimulates
angiogenesis. Endothelial cells coalesce to form rudimentary blood vessels, which subsequently
undergo maturation and stabilization. Simultaneously, fibroblasts begin to migrate and proliferate
within the wound bed and function to synthesize collagen and other components of the new
extracellular matrix.
III. Maturation
Wound contraction and collagen remodeling mark the
maturation phase of wound healing. Wound contraction is mediated
by myofibroblasts, which utilize specialized actin-myosin appendages
to contract the surrounding extracellular matrix. Over time,
equilibrium between collagen synthesis and degradation occurs. Type
I collagen is gradually replaced by type III collagen and extensive
cross-links develop to enhance the tensile strength of the scar to a
maximum value of 80% of non-wounded skin.
Figure 1
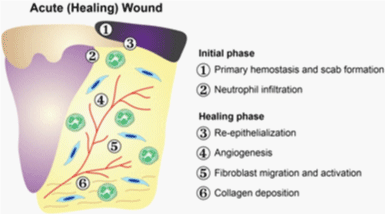
Figure 1
The acute wound healing process.
Figure 2
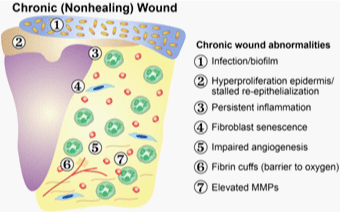
Figure 2
The chronic wound healing process.
Figure 3
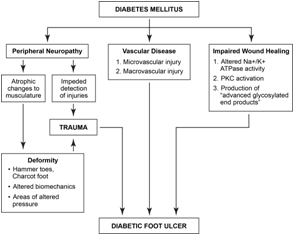
Figure 3
Pathophysiology of diabetic foot ulcers.
Deterrents to Physiologic Wound Healing
Several clinical factors affect the healing wound and can impede
or disrupt the wound-healing cascade. Important considerations
include hypoxia, infection, diabetes, malnutrition, and medications.
I. Hypoxia
Adequate tissue oxygenation is essential to physiologic wound
healing [2]. The wound-healing cascade will not proceed effectively
with tissue oxygen levels below 35 mmHg [3]. When tissue oxygen
levels fall below this critical value, the replication of fibroblasts is
impaired, ultimately resulting in deficient collagen production.
Furthermore, post-translational collagen hydroxylation is dependent
on molecular oxygen [2]. Numerous factors can result in tissue
hypoxia, including occlusive arterial disease, vasospastic disease,
vasculitis, hematologic disorders, and external compression of the
wound bed.
II. Infection
Bacterial contamination poses a significant impediment to
wound healing. Bacteria provide a constant antigenic stimulus,
which prolongs the inflammatory phase of wound healing. Chronic
inflammation results in the up-regulation of proteases, which degrade
growth factors and collagen within the local microenvironment
and inhibit further deposition of collagen, epithelialization, and
contraction of the wound [4].
III. Diabetes
The relationship between diabetes and impaired wound healing is
complex and research on that topic continues to evolve. Many factors
contribute to the altered wound healing seen in diabetic patients.
These include, but are not limited to, predisposition to other systemic
diseases such as atherosclerosis, renal failure with concomitant
uremia, peripheral arterial disease, and coagulopathy.
Uncontrolled hyperglycemia is thought to impede wound healing
on the molecular level in several different ways. Three hypotheses
seek to explain this phenomenon (Figure 3). The first hypothesis
involves the alteration of Na+/K+ ATPase activity [5]. Hyperglycemia
up-regulates the enzyme aldose reductase, which is responsible for
converting glucose to sorbitol via the poyol pathway. In this pathway,
sorbitol is then converted to fructose, resulting in the generation of
NADH. However, this enzymatic reaction is slow, thereby resulting
in a build-up of sorbitol within the cell. As an effective osmole, this
increases the osmotic stress on the cell and promotes swelling. In
addition, the increased conversion of sorbitol to fructose ultimately
causes reduction in NADPH levels, which makes the cells more
susceptible to oxidative stress. This is especially augmented by the
fact that chronic wounds have elevated levels of reactive oxidative
species. The second hypothesis involves activation of protein kinase C
(PKC) by hyperglycemia [6]. Diacyl-glycerol synthesis is elevated by
hyperglycemia, which, in turn, causes PKC activity to increase. PKC is
an important signaling molecule and alterations in its expression can
have detrimental effects on cellular proliferation, thereby impeding
wound healing. The third mechanism involves the production of
“advanced glycosylation end products” (AGEs) [7 and 8]. AGEs are
large aggregates of aldolases bound to reactive amino groups by
covalent bonds. These molecules are thought to activate NF-κB, a key
transcription factor involved in many cytokine-related cell responses.
AGEs may induce PDGF, TNF-α, and IL-1α and inhibit normal
collagen degradation.
IV. Malnutrition
Insufficient protein intake and vitamin/mineral deficiencies can
have profound consequences on wound healing. Dietary proteins are
the fundamental building blocks of collagen. Furthermore, protein
plays a key role in capillary formation, fibroblast proliferation, and
proteoglycan synthesis (GUO). While most agree that the normal
wound-healing cascade cannot precede with albumin levels less than 2
g/dL, impaired wound healing has been reported with albumin levels
less than 3.5 g/dL [9]. Protein replacement and supplementation can
reverse this process, hence the importance of early enteral feeds rich
in protein.
The most important vitamins for wound-healing are vitamins A,
C, E, and K. Vitamin A plays a fundamental role in the activation of
monocytes [10]. Vitamin C serves as a cofactor in the hydroxylation
of collagen residues and promotes proper immune function [11].
Vitamin E is the major anti-oxidant in skin and functions primarily
to stabilize cell membranes from reactive intermediates [4]. Vitamin
K is essential for the production of clotting factors necessary for
hemostasis (Factors VII, IX, X, prothrombin, Protein C, and Protein
S). In addition to vitamins, there are several important trace minerals
that are essential for proper wound healing, including zinc, iron,
copper, and magnesium. These elements are predominantly utilized
as cofactors for enzymatic reactions.
V. Medications
Numerous drugs impair the wound-healing process. While the
list continues to increase, chemotherapeutic agents, steroids, and
non-steroidal anti-inflammatory drugs (NSAIDs) are among the most
widely studied drugs that affect wound healing. Although different in
their respective mechanisms of action, this therapeutics ultimately
exerts their detrimental effects on wound healing by hampering the
inflammatory response. Impaired fibroblast proliferation results in
decreased collagen production, thereby preventing granulation tissue
formation and wound closure.
Common Chronic Wounds
I. Pressure ulcers
Pressure ulcers occur when a sustained mechanical force
compresses soft tissue for a prolonged period of time. The mechanism
of injury is complex and not completely understood, there is evidence
though that many factors come into play, including ischemia,
reperfusion injury, and impaired lymphatic drainage.
When an externally applied pressure, usually on a prominent
body surface, exceeds the capillary pressure within the tissue,
ischemia occurs. During ischemia, the cellular metabolism
transitions from aerobic to anaerobic, with deleterious effect to the
function of intracellular organelles and cell membrane. That leads
to cellular damage and promotes cellular necrosis. When the cause
of pressure is reversed, reperfusion of ischemic tissue can result
in the formation of reactive oxygen and nitrogen species, which
further damage surrounding tissue. Neutrophils are recruited to the
previously ischemic area and damage the surrounding tissue via the
release of inflammatory mediators, such as superoxide, hydroxyl
radicals, and peroxynitrite. Additionally, during periods of ischemia,
endothelial cells become activated to secrete pro-inflammatory
cytokines and increase the expression of adhesion molecules. All of
these components interact and eventually lead to tissue injury, which
becomes more extensive with repeated cycles of ischemia-reperfusion
[12].
Another mechanism, often underestimated, is the obstruction
of lymphatic drainage as a result of local pressure that leads to
inadequate removal of harmful waste products from the interstitial
space surrounding cells [13]. Lymph stasis has also been shown to
contribute to inflammation, fibrosis, and localized cell death, which
suggests that impaired lymphatic drainage plays a central role in the
pathophysiology of pressure ulcers.
Pressure ulcers, like all chronic wounds, undergo a prolonged
inflammatory phase. Microbial contamination is one of the main
factors contributing to that. A biofilm often develops, which
consistently activates the immune system. Chemotactic factors such
as IL-1 and TNF-α are produced, attracting immune cells including
macrophages, neutrophils, and mast cells. Reactive oxygen species
(ROS), neutrophil elastase, and matrix metalloproteinases (MMPs)
are subsequently produced. The protective glycocalyx of the biofilm
prevents penetration of bactericidal molecules, protecting the
underlying bacteria and perpetuating the chronic contamination
and potentially infection of the wound, preventing proper wound
healing. The function of fibroblasts is also impaired secondary to
altered response to growth factors, which has a detrimental effect
on the proliferative phase of wound healing. Moreover, inhibition
of signaling pathways responsible for keratinocyte migration, lead
to impaired keratinocyte translocation from the wound edges, which
further impairs wound healing.
From a clinical perspective, most pressure ulcers occur in
patients who demonstrate a number of intrinsic risk factors including
advanced age, medications (i.e., corticosteroids, vasoactive agents),
comorbid diseases (cancer, cardiovascular disease, peripheral vascular
disease, diabetes), anemia, low systolic or mean blood pressure, body
mass index (BMI) less than 18.5 or greater than 40, poor nutritional
status and low albumin levels. These factors affect the ability of the
skin to respond to extrinsic risk factors that include shear, friction,
moisture and pressure.
A full physical exam and history is important for evaluation of
patients with pressure ulcers. Important considerations include
social history, spasms management, and bladder and bowel habits.
Control of spasms is critical as they can lead to shearing forces that
impair wound healing. They can be managed either medically (e.g.,
Botox, Baclofen, Diazepam, Dantrolene, Gabapentin, Tizanidine) or
surgically (e.g., Baclofen pumps, spinal electric stimulation therapy,
nerve blocks, surgical rhizotomy, joint releases). Additionally,
optimization of patient’s nutrition and medical condition is very
important. For diabetic patients, it is critical to achieve tight blood
glucose control with HbA1c below 7%. All pressure sores are
considered colonized since they are open wounds; however, it is
critical to determine the evidence of active and invasive infection.
Imaging studies, such as CT scan, bone scan, MRI, can be helpful, but
the gold standard to assess for soft tissue infection is tissue biopsy with
quantitative culture. Bacterial count greater than 105 is considered
diagnostic for invasive infection and needs to be addressed in order to
increase the chances of a successful reconstruction. If infection of the
bone is suspected, bone biopsy is required to rule out osteomyelitis.
Occasionally, pulmonary and urinary sources cause transient
bacteremia and seeding of the bloodstream with potential subsequent
infection of the pressure ulcers. Treatment of any other sources
of infection is equally important to treating the pressure wound
infection. For incontinent patients, any pelvic, gluteal or lower back
wounds are potentially continuously exposed to urine and/or feces,
which can make management of these wounds more challenging.
Discussion with the patient for potential diverting colostomy may be
recommended, as it significantly increases the chances of successful
pressure sore treatment.
Depending on the depth of the ulcer and the level of involvement,
pressure sores are classified in 4 stages. In stage I ulcers, the skin is
intact with areas of non-blanchable erythema. In stage II ulcers, there
is evidence of partial-thickness skin loss with exposed dermis. The
wound bed is viable, pink or red and moist. They may appear as intact
or ruptured serum-filled blisters. Adipose and deeper tissues are not
visible and granulation tissue, slough and eschar are not present.
In stage III ulcers, there is full-thickness loss of skin with exposed
subcutaneous adipose tissue and evidence of granulation tissue and
epibole (rolled wound edges). Finally, in stage IV ulcers there is fullthickness
skin and tissue loss with exposed fascia, muscle, tendon,
ligament, cartilage and/or bone. Slough, eschar, epibole, undermining
and/or tunneling may be present. If slough or eschar is covering
the wound and obscures the extent of tissue loss, this is defined as
unstageable pressure ulcer. Representative images of pressure sores
are shown in Figures 4-6.
The size and depth of the wound, presence of exposed structures,
especially tendons, bursa or bone, neurovascular structures, and the
relationship of the wound to underlying or adjacent bony prominences,
as well as the rectum and urethra, should be assessed thoroughly.
Pressure ulcers can be treated non-operatively by promoting
secondary intention healing. A combination of interventions is
utilized, including turning the patient every 2 hours, keeping bed
linens clean, dry and wrinkle free, using soft cushions between knees
and boney prominences to avoid direct contact, moving immobile
patient with caution. In addition, cleaning and protecting the skin
surrounding the pressure sore to prevent further break down,
relieving pressure, friction, shear, and moisture, supplementing
patient’s diet with multivitamin, zinc sulfate, and Vitamin C, using
low air loss mattress and performing appropriate dressing changes.
Split or full thickness skin grafts are alternative reconstruction
modalities when secondary healing is unlikely to achieve complete
closure of the wound in a timely fashion. In case major reconstruction
is indicated, local flaps can be considered, although fasciocutaneous
or myocutaneous flaps are the gold standard for treatment of nonhealing,
large wounds that involve deeper structures, such as bone,
bursa, muscle or tendons.
II. Diabetic foot ulcers
Diabetic foot ulcers are one of the most serious manifestations
of uncontrolled diabetes with a reported incidence of 4 to 10%
[14]. Diabetic foot ulcers are a source of considerable morbidity
and mortality, including discomfort, decreased quality of life, need
for health care provider visits, wound care, and need for surgical
intervention [15].
Diabetic foot ulcers are the culmination of several distinct
underlying pathophysiological processes. Previous studies have
shown that the majority of patients with diabetic foot ulcers have
a critical triad of peripheral neuropathy, deformity, and trauma
(Figure 3) [16]. Peripheral neuropathy is considered the earliest
feature in the development of diabetic foot ulcers [15]. Motor
neuropathy promotes atrophic changes of the foot musculature,
ultimately resulting in characteristic deformities of the feet, including
hammertoes and Charcot arthropathy. The altered biomechanics
of the foot create focal areas of pressure that are prone to injury.
Furthermore, altered sensation secondary to neuropathy impedes
the initial detection of injuries and allows progression of the wounds
when they occur. Coexisting peripheral vascular disease also
contributes to the pathogenesis of diabetic foot ulcers. Over time,
non-enzymatic glycosylation of the endothelium causes sclerosis of
the vessel wall. Diminished perfusion contributes to hypoxia and
ischemic necrosis of the affected tissue. Uncontrolled hyperglycemia
results in impaired leukocyte chemotaxis, phagocytosis, and killing,
thus increasing susceptibility to microbial infection. Furthermore,
the relative immunodeficiency impedes the inflammatory phase of
wound healing and diminishes the regenerative capacity of the tissue.
The clinical evaluation of diabetic foot ulcers begins with a complete
history and physical, including a systematic assessment of the lower
extremities. Vascular status should be assessed by the palpation of
the dorsalis pedis and posterior tibial pulses. Neurological assessment
should be conducted using a monofilament. Inability to detect a 10-g
monofilament has been shown to convey a 2.2-fold to 18-fold risk of
ulceration [16]. Further neurological testing includes assessment of
deep tendon reflexes at the ankle. The foot should then be inspected
for gross deformity. The patient’s footwear should be examined.
Important considerations include excessively worn shoes, shoes with
obvious weakness in the sole, and the presence of stitches or seams
around the toes [17]. The location of the ulcers should be noted and
assessed for diameter and depth. Given the impaired immunologic
function of diabetic patients with uncontrolled hyperglycemia, the
wounds may lack the characteristic signs of infection such as warmth,
redness, and swelling. Wounds, which extend all the way down to the
bone, have a high predictive value for underlying osteomyelitis, even
without acute signs of infection (Figure. 7-9) [16].
There are currently several clinically utilized classification
systems for diabetic foot ulcers. The two most commonly used
classification systems include the Wagner system and the University
of Texas system [14,18-20]. The Wagner system assesses the depth
of the diabetic foot ulcer and the presence of osteomyelitis and
gangrene (Table 1) [18,19]. The University of Texas system assesses
ulcer depth, the presence of wound infection, and the presence of
clinical signs of ischemia [19,20]. Recently, a new diabetic foot ulcer
assessment scale (DFUAS) was developed in Indonesia. This newly
developed system consists of 11 evaluation components (depth, size,
size score, inflammation/infection, proportion of granulation tissue,
type of necrotic tissue, proportion of necrotic tissue, proportion of
slough, maceration, type of wound edge, and tunneling) which are
assessed to yield an overall score ranging from 0 to 98, with higher
scores corresponding to more severe wounds [14]. Using this system,
the authors of the study determined that a cutoff score of 12 could
accurately predict whether a wound would heal within 4 weeks [14].
The clinical management of diabetic foot ulcers should begin with
patient education. Patients should be taught about the underlying
pathology of diabetic foot ulcers in terms that are comprehensible for
their corresponding level of education. The importance of glycemic
control should be emphasized. Conservative management options
include rest, elevation, and pressure off-loading of the ulcer. Offloading
the ulcer can be accomplished in several different ways,
including changing the patient’s footwear, creating a complex molded
device, or utilizing an aircast boot [17]. Total contact casts have been
regarded as a superior standard therapy in neuropathic ulcers due to
the purported ability to redistribute pressure, decrease patient activity
levels, and increase adherence to the off-loading regiment [16].
Debridement is the mainstay of ulcer therapy. Callus surrounding the
foot ulcer impedes wound healing by preventing epithelial migration
across the bed of the wound [17]. Necrotic tissue is no longer viable
and serves as a nidus for infection. Thus, these tissues should be
removed in order to promote physiologic wound healing. Tissue
samples should be taken and sent for further pathologic and infectious
work-up. Antibiotics should be administered to combat infection
when present and tailored to the infective organism. It is imperative to
keep the affected area clean with sterile and nonadhesive dressings. In
cases refractory to conservative management, more extensive surgical
debridement, including potential amputation, should be considered.
III. Venous ulcers
Ulceration secondary to chronic venous insufficiency is a
common problem in the United States, with an estimated prevalence
of 1-2% in the adult population [21]. Chronic venous insufficiency is
more common among those who are obese, pregnant, and sedentary,
have incurred trauma to the vasculature secondary to prior injury or
surgery, or have a family history of varicose veins.
Venous insufficiency is multifactorial in nature and can result from
several different causes, including; valvular incompetence, outflow
obstruction, congenital weakness of the vasculature, or ineffective
pumping of the surrounding musculature [21]. Regardless of the
underlying cause, this ultimately results in pooling of blood within
the venous system. The resulting hypertension leads to extravasation
of multiple blood proteins, most notably hemosiderin and fibrinogen.
Hemosiderin deposition results in the hyperpigmentation of the
overlying skin. Fibrinogen is believed to polymerize into fibrin cuffs,
which impede oxygen diffusion and entrap growth factors, thereby
promoting ischemic necrosis and ulceration of the overlying skin.
Patients with venous stasis commonly present with complains
of a dull ache or pain in the affected area [22]. On physical exam,
the skin will often have a characteristic dark pigmentation resulting
from hemosiderin deposition. Other associated skin findings include
lipodermatosclerosis, reticular or varicose veins, atrophic Blanche,
telangiectasia, and stasis eczema [23]. Ulcers often form above the
medial malleolus and tend to be superficial with poorly defined
margins (Figure 10,11) [23].
Non-operative treatment of venous ulcers is centered on
compression therapy and dressing changes to prevent concomitant
infection. There are several different clinically available options for
compression therapy, including bandages, stockings, and boots
[24]. In addition to standard wound care; several pharmacological
therapies are available to attenuate the inflammation associated with
venous ulceration. Examples of such therapies include pentoxifylline,
an inhibitor of adenylate cyclase, and micronized purified flavonoid
fraction (MPFF). Artificial skin substitutes and autologous skin
grafting can be used in cases refractory to medical management.
Figure 4
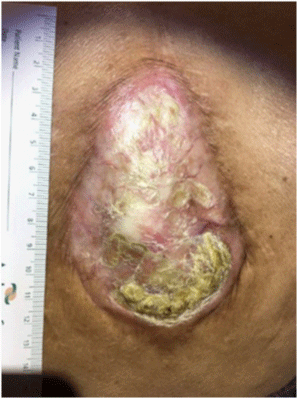
Figure 4
Lower back, lumbar area pressure ulcer with complete reepithelialization
after medical optimization and appropriate wound care.
Figure 5
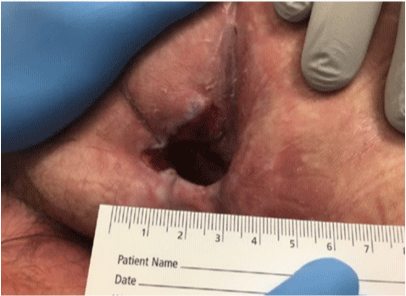
Figure 5
Sacral pressure ulcer, stage IV; clear wound edges with no drainage or sign of infection, presence of granulation tissue at the base of the wound.
Figure 6
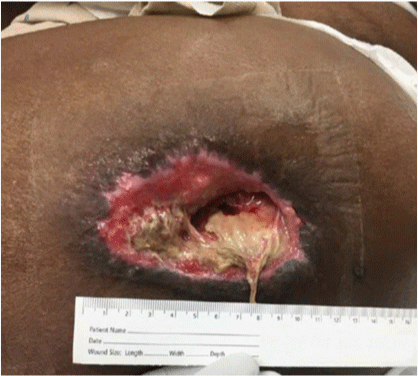
Figure 6
Trochanteric pressure ulcer stage IV; moderate amount of drainage and fibrinous tissue covering part of the wound but no signs of infection, evidence of granulation tissue with small amount of epibole at the wound edges.
Figure 7
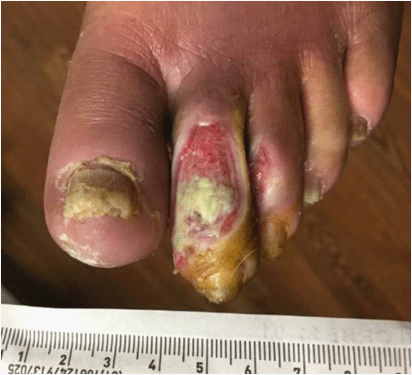
Figure 7
Diabetic ulcer of the foot; full thickness defect with evidence of
granulation tissue and small area of fibrinous exudate.
Figure 8
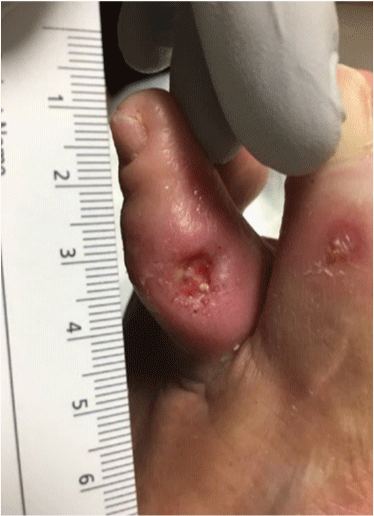
Figure 8
Diabetic ulcer of the 2nd toe; healing wound with granulation tissue
and no evidence of infection.
Figure 9
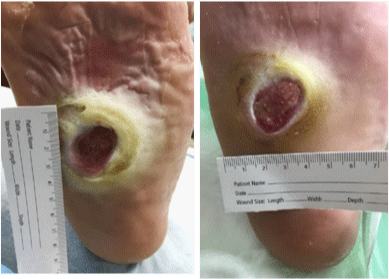
Figure 9
Diabetic ulcer of the plantar surface of the foot; full thickness wound with evidence of granulation tissue and significant amount of hyperkeratosis, pictures before and after local wound care and limited bedside debridement.
Table 1
Table 1
Wagner system for classification of diabetic foot wounds.
Figure 10
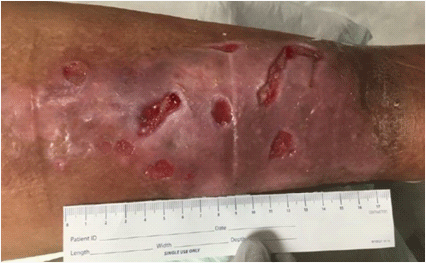
Figure 10
10: Venous stasis ulcers; evidence of pitting edema, hyperpigmentation and full thickness wounds with granulation tissue and small amount of exudate.
Figure 11
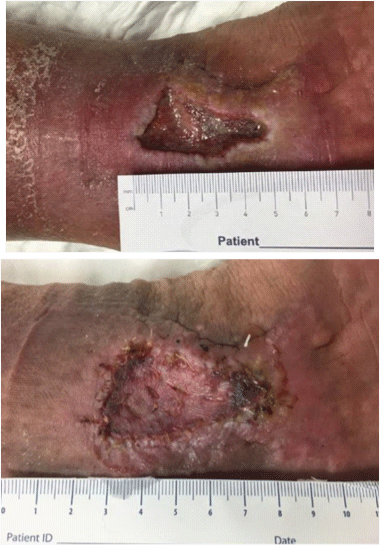
Figure 11
Venous stasis ulcer at the medial malleolus; pictures before and after split thickness skin graft placement.
Hyperbaric Oxygen Therapy
Hyperbaric oxygen therapy (HBOT) is another modality used in the treatment of specific types of wounds. The patient is placed in a chamber that has the ability to deliver up to 100% oxygen. That increases the delivery of oxygen to areas with poor perfusion or increased metabolic demand because of inflammation and/or infection, improving the wound healing process. HBOT is currently indicated for the treatment of chronic diabetic and radiation wounds, as well as chronic wounds with evidence of osteomyelitis [25-27]. Potential complications, such as barotrauma, oxygen toxicity and visual changes, need to be carefully considered. Additionally, it is important to mention that the cost of HBOT is high, requiring expensive equipment and specialized staff. However, there is literature that supports the cost-effectiveness of HBOT in a multidisciplinary wound care center setting with appropriate utilization of the available resources [25,26].
Future Horizons for Healing of Chronic Wounds
Advances in the field of regenerative medicine have led to the
development of novel therapeutic modalities, which seek to accelerate
physiologic wound healing and improve cosmetic outcomes. Many
of these therapies aim to augment and manipulate factors within the
wound bed in order to favor a pro-regenerative microenvironment.
The extracellular matrix is critically important to the physiologic
healing of wounds and has been a target of substantial scientific
investigation. Both natural and synthetic biomaterials are currently
being developed to guide wound regeneration. Ideally, these materials
will guide the production of new host extracellular matrix while being
simultaneously degraded into non-toxic byproducts. Current options
include acellular dermal matrix (ADM) and polyesters made of
polycaprolactone, polyglycolide, and polylactic acid [28].
Acellular dermal matrices are composed of several different
components, including collagen, glycoproteins, glycosaminoglycans,
adhesion molecules, growth factors, chemokines, and cytokines. The
complex architecture of the acellular dermal matrix helps to guide
regeneration in part by promoting a phenotypic change of M1-like,
pro-inflammatory macrophages to M2-like pro-healing macrophages
[29]. Furthermore, because ADM is natural, it is metabolized into
non-toxic degradation products which have been shown to promote
wound regeneration and facilitate cellular recruitment [27]. The
efficacy of acellular dermal matrices has been verified in a randomized
control trial for both diabetic foot ulcers [30].
Synthetic polymers offer the additional benefits of being able to
alter the physiochemical properties of the biomaterial as well as load
the material with bioactive molecules, which promote regeneration.
Synthetic scaffolds can be produced by several different manufacturing
processes, including electrospinning to produce nano-fiber scaffolds,
salt-leaching to produce porous scaffolds, and lithography to produce
three-dimensional scaffold [28]. By altering the chemical composition
of the polymer, one can augment the degradation kinetics, mechanical
strength, stiffness, porosity, and permeability. Moreover, there have
been several studies, which demonstrate that loading these polymers
with cytokines, growth factors, siRNA, and genes can promote a proregenerative
environment [31-36].
Stem cells provide another potential therapeutic strategy to
improve cutaneous wound healing. Through self-renewal and
daughter cell differentiation, stem cells replenish damaged tissues
directly through proliferation and by secreting a host of proregenerative
factors. Although there are several different types of
stem cells that can be utilized for wound healing applications, adipose
derived stem cells tend to be the preferred subtype given the ease at
which adipose tissue can be harvested and the relative density of stem
cells within this tissue. Although the efficacy of utilizing adiposestem
cells to enhance wound healing has been documented in several
studies, this technology has yet to be translated clinically [37,38].
Multidisciplinary Approach in Wound Care
The increasing need for care of non-healing wounds, combined with the great advances in the science of wound care have led to the development of specialized multidisciplinary teams and centers. These teams usually comprise of physicians, nurses and technicians from different specialties, including plastic surgery, vascular surgery, general surgery, podiatry, dermatology and hyperbaric medicine. It has been shown that multidisciplinary approach can improve the healing rates of chronic wounds, decrease hospitalizations and reduce the rate of complications related to non-healing wounds [39 and 40]. For instance, our institution’s wound care center provides advanced conventional wound care, such as application of specialized dressings and wound debridement, compression therapy for venous stasis ulcers, surgical and non-surgical techniques for wound closure, including vacuum-assisted technology, tissue replacement with skin grafts or flaps, skin substitutes or scaffolds and HBOT. More evidencebased research is required in order to develop validated guidelines for the function of multidisciplinary wound care centers [41].
Conclusion
Healing chronic wounds presents a formidable challenge to the field of plastic and reconstructive surgery. Patients with non-healing wounds often have multiple comorbidities, which further complicate their medical care. A thorough understanding of the underlying principles of wound healing and available treatment modalities is essential to optimizing the care of these patients. Future efforts will further expand on the clinical armamentarium available to reconstructive surgeons to help rebuild and restore the wounds and lives of the patients we serve.
References
- Phillips LG. Wound healing. In: Townsend CM Jr., Beauchamp RD, Evers BM, Mattox KL, editors. Sabiston Textbook of Surgery. Philadelphia: W.B. Saunders. 2001; 131-144.
- Sen CK. Wound healing essentials: let there be oxygen. Wound Repair Regen. 2009; 17: 1-18.
- Knighton DR, Hunt TK, Scheuenstuhl H, Halliday BJ, Werb Z, Banda MJ. Oxygen tension regulates the expression of angiogenesis factor by macrophages. Science. 1983; 221: 1283-1285.
- Guo S, Dipietro LA. Factors affecting wound healing. J Dent Res. 2010; 89: 219-229.
- Greene DA, Lattimer SA, Sima AA. Sorbitol, phosphoinositides, and sodium-potassium-ATPase in the pathogenesis of diabetic complications. N Engl J Med. 1987; 316: 599-606.
- Lee TS, Saltsman KA, Ohashi H, King GL. Activation of protein kinase C by elevation of glucose concentration: proposal for a mechanism in the development of diabetic vascular complications. Proc Natl Acad Sci U S A. 1989; 86: 5141-5145.
- Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med. 1988; 318: 1315-1321.
- Yan SD, Schmidt AM, Anderson GM, Zhang J, Brett J, Zou YS, et al. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem. 1994; 269: 9889-9897.
- Bohl DD, Shen MR, Mayo BC, Massel DH, Long WW, Modi KD, et al. Malnutrition Predicts Infectious and Wound Complications Following Posterior Lumbar Spinal Fusion. Spine (Phila Pa 1976). 2016;
- Ross AC. Vitamin A and protective immunity. Nutr Today. 1992; 27: 18- 26.
- Hunt A. The role of vitamin C in wound healing. Br J Surg. 1940; 28: 436- 461.
- Peirce SM, Skalak TC, Rodeheaver GT. Ischemia-reperfusion injury in chronic pressure ulcer formation: a skin model in the rat. Wound Repair Regen. 2000; 8: 68-76.
- Gray RJ, Voegeli D, Bader DL. Features of lymphatic dysfunction in compressed skin tissues - Implications in pressure ulcer aetiology. J Tissue Viability. 2016; 25: 26-31.
- Arisandi D, Oe M, Roselyne Yotsu R, Matsumoto M, Ogai K, Nakagami G, et al. Evaluation of validity of the new diabetic foot ulcer assessment scale in Indonesia. Wound Repair Regen. 2016. [epub ahead of print.]
- Noor S, Khan RU, Ahmad J. Understanding diabetic foot infection and its management. Diabetes Metab Syndr. 2016; S1871-4021.
- Frykberg RG, Banks J. Management of diabetic foot ulcers: A review. Current Psychiatry. 2016; 33: 16-23.
- Roberts P, Newton V. Assessment and management of diabetic foot ulcers. Br J Community Nurs. 2011; 16: 485-486, 488-490.
- Wagner FJ. A classification and treatment program for diabetic, neuropathic and dysvascular foot problems. Instr Course Lect. 1979; 28: 143–165.
- Oyibo SO, Jude EB, Tarawneh I, Nguyen HC, Harkless LB, Boulton AJ. A comparison of two diabetic foot ulcer classification systems: the Wagner and the University of Texas wound classification systems. Diabetes Care. 2001; 24: 84-88.
- Armstrong DG, Lavery LA, Harkless LB. Validation of a diabetic wound classification system. The contribution of depth, infection, and ischemia to risk of amputation. Diabetes Care. 1998; 21: 855-859.
- Alavi A, Sibbald RG, Phillips TJ, Miller OF, Margolis DJ, Marston W, et al. What's new: Management of venous leg ulcers: Approach to venous leg ulcers. J Am Acad Dermatol. 2016; 74: 627-640.
- Collins L, Seraj S. Diagnosis and treatment of venous ulcers. Am Fam Physician. 2010; 81: 989-996.
- Spentzouris G, Labropoulos N. The evaluation of lower-extremity ulcers. Semin Intervent Radiol. 2009; 26: 286-295.
- Pascarella L, Shortell CK. Medical management of venous ulcers. Semin Vasc Surg. 2015; 28: 21-28.
- Kranke P, Bennett M, Roeckl-Wiedmann I, Debus S. Hyperbaric oxygen therapy for chronic wounds. Cochrane Database Syst Rev. 2004: CD004123.
- Abidia A, Laden G, Kuhan G, Johnson BF, Wilkinson AR, Renwick PM, et al. The role of hyperbaric oxygen therapy in ischaemic diabetic lower extremity ulcers: a double-blind randomised-controlled trial. Eur J Vasc Endovasc Surg. 2003; 25: 513-518.
- Kalani M, Jorneskog G, Naderi N, Lind F, Brismar K. Hyperbaric oxygen (HBO) therapy in treatment of diabetic foot ulcers. Long-term follow-up. J Diabetes Complications. 2002; 16: 153-158.
- Hu MS, Maan ZN, Wu JC, Rennert RC, Hong WX, Lai TS, et al. Tissue engineering and regenerative repair in wound healing. Ann Biomed Eng. 2014; 42: 1494-1507.
- Swinehart IT, Badylak SF. Extracellular matrix bioscaffolds in tissue remodeling and morphogenesis. Dev Dyn. 2016; 245: 351-360.
- Zelen CM, Orgill DP, Serena T, Galiano R, Carter MJ, DiDomenico LA, et al. A prospective, randomised, controlled, multicentre clinical trial examining healing rates, safety and cost to closure of an acellular reticular allogenic human dermis versus standard of care in the treatment of chronic diabetic foot ulcers. Int Wound J. 2016.
- Mittermayr R, Slezak P, Haffner N, Smolen D, Hartinger J, Hofmann A, et al. Controlled release of fibrin matrix-conjugated platelet derived growth factor improves ischemic tissue regeneration by functional angiogenesis. Acta Biomater. 2016; 29: 11-20.
- Li W, Lan Y, Guo R, Zhang Y, Xue W, Zhang Y. In vitro and in vivo evaluation of a novel collagen/cellulose nanocrystals scaffold for achieving the sustained release of basic fibroblast growth factor. J Biomater Appl. 2015; 29: 882-893.
- Lai HJ, Kuan CH, Wu HC, Tsai JC, Chen TM, Hsieh DJ, et al. Tailored design of electrospun composite nanofibers with staged release of multiple angiogenic growth factors for chronic wound healing. Acta Biomater. 2014; 10: 4156-4166.
- Freudenberg U, Zieris A, Chwalek K, Tsurkan MV, Maitz MF, Atallah P, et al. Heparin desulfation modulates VEGF release and angiogenesis in diabetic wounds. J Control Release. 2015; 220: 79-88.
- Kim HS, Son YJ, Yoo HS. Clustering siRNA conjugates for MMPresponsive therapeutics in chronic wounds of diabetic animals. Nanoscale. 2016; 8: 13236-13244.
- Kwon MJ, An S, Choi S, Nam K, Jung HS, Yoon CS, et al. Effective healing of diabetic skin wounds by using nonviral gene therapy based on minicircle vascular endothelial growth factor DNA and a cationic dendrimer. J Gene Med. 2012; 14: 272-278.
- Strong AL, Bowles AC, MacCrimmon CP, Frazier TP, Lee SJ, Wu X, et al. Adipose stromal cells repair pressure ulcers in both young and elderly mice: potential role of adipogenesis in skin repair. Stem Cells Transl Med. 2015; 4: 632-642.
- Yoshida S, Yoshimoto H, Hirano A, Akita S. Wound Healing and Angiogenesis through Combined Use of a Vascularized Tissue Flap and Adipose-Derived Stem Cells in a Rat Hindlimb Irradiated Ischemia Model. Plast Reconstr Surg. 2016; 137: 1486-1497.
- Sholar AD, Wong LK, Culpepper JW, Sargent LA. The specialized wound care center: a 7-year experience at a tertiary care hospital. Ann Plast Surg. 2007; 58: 279-284.
- Almdal T, Nielsen AA, Nielsen KE, Jørgensen ME, Rasmussen A, Hangaard S, et al. Increased healing in diabetic toe ulcers in a multidisciplinary foot clinic-An observational cohort study. Diabetes Res Clin Pract. 2015; 110: 315-321.
- Warriner RA 3rd, Carter MJ. The current state of evidence-based protocols in wound care. Plast Reconstr Surg. 2011; 127: 144S-153S.